| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://ijcp.elmerpub.com |
Original Article
Volume 15, Number 1, March 2026, pages 1-7

Complications of Autosomal Dominant Polycystic Kidney Disease in Pediatrics: A Twenty-Five-Year Experience
Mariana Costaa, d ![]() , Joao Diasb, Marta Machadoc, Catarina Nevesc, Carmen do Carmoc, Carolina Cordinhac, Clara Gomesc
, Joao Diasb, Marta Machadoc, Catarina Nevesc, Carmen do Carmoc, Carolina Cordinhac, Clara Gomesc
aCoimbra Pediatric Hospital, Local Health Unit (ULS) of Coimbra, Coimbra, Portugal
bPediatric Cardiology Department, Coimbra Pediatric Hospital, Local Health Unit (ULS) of Coimbra, Portugal
cPediatric Nephrology Unit, Coimbra Pediatric Hospital, Local Health Unit (ULS) of Coimbra, Portugal
dCorresponding Author: Mariana Costa, Coimbra Pediatric Hospital, Local Health Unit (ULS) of Coimbra, Coimbra, Portugal
Manuscript submitted January 12, 2026, accepted March 10, 2026, published online March 26, 2026
Short title: Complications of ADPKD in Pediatrics
doi: https://doi.org/10.14740/ijcp1041
| Abstract | ▴Top |
Background: Autosomal dominant polycystic kidney disease (ADPKD) is a systemic ciliopathy caused by pathogenic variants in PKD1 or PKD2, predominantly affecting the kidneys. Although traditionally considered as an adult-onset condition, ADPKD may present during childhood, with early manifestations such as proteinuria and hypertension. However, predictors of early complications in pediatric ADPKD remain limited. This study aimed to characterize the clinical manifestations of pediatric ADPKD and to explore potential early predictors of disease-related complications.
Methods: We conducted a retrospective descriptive study of pediatric patients (< 18 years) with ADPKD followed at a tertiary center between 1999 and 2024. Clinical data included demographics, family history, estimated glomerular filtration rate (eGFR), ultrasonographic parameters, and ADPKD-related complications, according to Kidney Disease: Improving Global Outcomes (KDIGO) 2025 guidance.
Results: Sixty-seven children were included (52.2% male), with a mean age at first evaluation of 7.4 ± 5.5 years. Twelve patients (17.9%) were diagnosed before 2 years of age, and prenatal renal cystic abnormalities were documented in six (9.0%). Family history was present in 97.0%. The mean kidney length Z-score was 1.94 ± 2.43; 59.7% had ≥ 10 cysts, and 41.8% had Bosniak category II or higher. Mean eGFR was 97.9 ± 15.1 mL/min/1.73 m2, and 22.2% had chronic kidney disease stage II. During follow-up (mean 4.0 ± 4.3 years), 27.9% developed complications, most commonly urinary tract infections, proteinuria, and hypertension. Complications were more frequent in females, while males had lower eGFR. No associations were found between complications and imaging parameters.
Conclusions: ADPKD is associated with a substantial burden of complications in childhood. In the absence of validated prognostic markers or disease-modifying therapies for pediatric patients, early recognition and structured follow-up remain essential.
Keywords: Children; Hypertension; Pediatric nephrology; Polycystic kidney disease; Renal cysts
| Introduction | ▴Top |
Autosomal dominant polycystic kidney disease (ADPKD) is a systemic ciliopathy caused by pathogenic variants in the PKD1 or PKD2 genes and predominantly affects the kidneys [1–3]. With an estimated incidence of 3.96 per 10,000 individuals, ADPKD has traditionally been considered as an adult-onset disease. However, growing evidence, reinforced by the 2025 Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, indicates that clinically relevant manifestations may already occur during childhood [1–3]. Pediatric studies have shown that children with ADPKD may present early complications, particularly proteinuria and hypertension, reported in up to 30% of cases [4, 5]. Additionally, early cardiovascular and metabolic alterations have been described, such as increased arterial stiffness, endothelial dysfunction, low-grade systemic inflammation, and subtle cardiac structural changes, suggesting early cardiometabolic involvement, namely alterations in lipid parameters and biomarkers of oxidative stress [6].
Because simple kidney cysts are uncommon in children, ADPKD is usually suspected based on imaging findings in the presence of a positive family history. According to KDIGO 2025, a presumptive diagnosis can be established in an at-risk child with at least one renal cyst [1]. Prenatal suspicion may arise from fetal ultrasound findings in fetuses with an affected parent, while genetic testing provides definitive diagnosis and is recommended for atypical cases, in the absence of family history, or when imaging findings are inconclusive. In pediatric patients, the decision to pursue genetic testing requires careful consideration of both diagnostic benefit and ethical implications [1].
Several clinical complications, such as hypertension, proteinuria, and macroscopic hematuria have been associated with a faster renal function decline in ADPKD [7, 8]. In children, correlations between kidney size, cyst burden, and elevated ambulatory blood pressure have been reported [9]. Nevertheless, unlike in adults, where prognostic tools such as the PROPKD score are available [10], no validated prognostic model exists for pediatric ADPKD.
The aim of this study was to characterize the clinical manifestations of ADPKD in a pediatric population and to explore potential early predictors of complications in this age group.
| Materials and Methods | ▴Top |
We conducted a retrospective descriptive study based on the analysis of clinical data from all individuals under 18 years of age diagnosed with ADPKD, who were followed at a tertiary referral hospital in central Portugal, between January 1999 and December 2024. Data collected included demographic characteristics, height and weight percentiles, family history, and renal function by estimated glomerular filtration rate (eGFR) using the modified Schwartz equation. Ultrasonographic parameters were also recorded, including kidney length converted to age-adjusted Z-scores using published pediatric reference data (14), total number of cysts, largest cyst diameter, and cyst complexity, classified according to the Bosniak system.
Disease-related clinical manifestations considered in this study were predefined as urinary tract infections, non-nephrotic proteinuria, hypertension (clinic measurements ≥ 95th percentile for age, sex, and height), enuresis or nocturia, nephrolithiasis, and macroscopic hematuria.
Statistical analysis was performed using IBM SPSS Statistics 24®, and statistical significance was defined as P < 0.05.
Institutional review board approval
According to national legislation and institutional policy, this retrospective study using fully anonymized clinical data was exempt from institutional review board approval.
Ethical compliance
This retrospective study was conducted using fully anonymized clinical data collected as part of routine clinical care and was performed in accordance with the ethical principles of the Declaration of Helsinki. According to national legislation and institutional policy, ethics committee approval was not required.
| Results | ▴Top |
Sociodemographic data
Sixty-seven pediatric patients with either a presumed diagnosis of ADPKD (defined by the presence of an affected parent and at least one renal cyst) or a genetically confirmed diagnosis were included (52.2% male). A family history of ADPKD was present in 97.0% of cases, with maternal transmission in 61.2%. The mean age at first evaluation was 7.4 ± 5.5 years. At the time of data collection, the mean age was 9.5 ± 5.9 years for males and 14.4 ± 4.5 years for females. Twelve children (17.9%) were diagnosed within the first 2 years of life. Prenatal renal cystic abnormalities compatible with a subsequent diagnosis of ADPKD were documented in six patients (9.0%).
Ultrasonographic findings and renal function
Renal ultrasound revealed a mean kidney length Z-score of 1.94 ± 2.43. Ten or more renal cysts were observed in 59.7% of patients. Cyst complexity was classified as Bosniak category II or higher in 41.8% of the cohort, including 3.0% with category IIF lesions. Renal function data were available for 54 patients (78%), with a mean eGFR of 97.9 ± 15.1 mL/min/1.73 m2. Among these patients, 12 (22.2%) met criteria for chronic kidney disease stage II.
Clinical manifestations
Overall, 19 of the 67 patients (28.4%) developed at least one ADPKD-related clinical manifestation during follow-up (mean duration 4.0 ± 4.3 years). Among the 54 patients with available eGFR data, 17 (31.5%) experienced complications.
The most frequent complications were urinary tract infections (11.9%), non-nephrotic proteinuria (8.9%), hypertension (8.9%), enuresis or nocturia (7.5%), nephrolithiasis (4.5%), and macroscopic hematuria (1.5%). Children who developed complications were significantly older than those without complications (14.2 ± 5.2 vs. 10.9 ± 5.0 years; P = 0.015). Overall, complications occurred more frequently in females than in males (43.8% vs. 14.3%; P = 0.008). In contrast, male patients had significantly lower eGFR values compared with females (93.0 ± 16.1 vs. 103.1 ± 15.6 mL/min/1.73 m2; P = 0.015). No association was observed between the presence of complications and eGFR decline. Sex- and age-related differences and their association with renal function are summarized in Table 1.
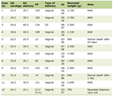 Click to view | Table 1. Demographic, Clinical, and Imaging Characteristics of Pediatric Patients With ADPKD |
Predictive factors
No significant associations were identified between the occurrence of complications and weight percentile, kidney dimensions, cyst number, cyst complexity, or largest cyst diameter. The distribution of kidney length Z-scores in patients with and without complications is shown in Figure 1. Ultrasonographic parameters were not associated with differences in eGFR. The relationship between kidney length Z-score and eGFR is illustrated in Figure 2. Linear regression analysis evaluating the association between age and eGFR is presented in Figure 3, showing no significant correlation. Similarly, the distribution of largest cyst diameter according to the presence of complications is illustrated in Figure 4. Finally, eGFR values according to cyst complexity (Bosniak categories II and IIF) are presented in Figure 5.
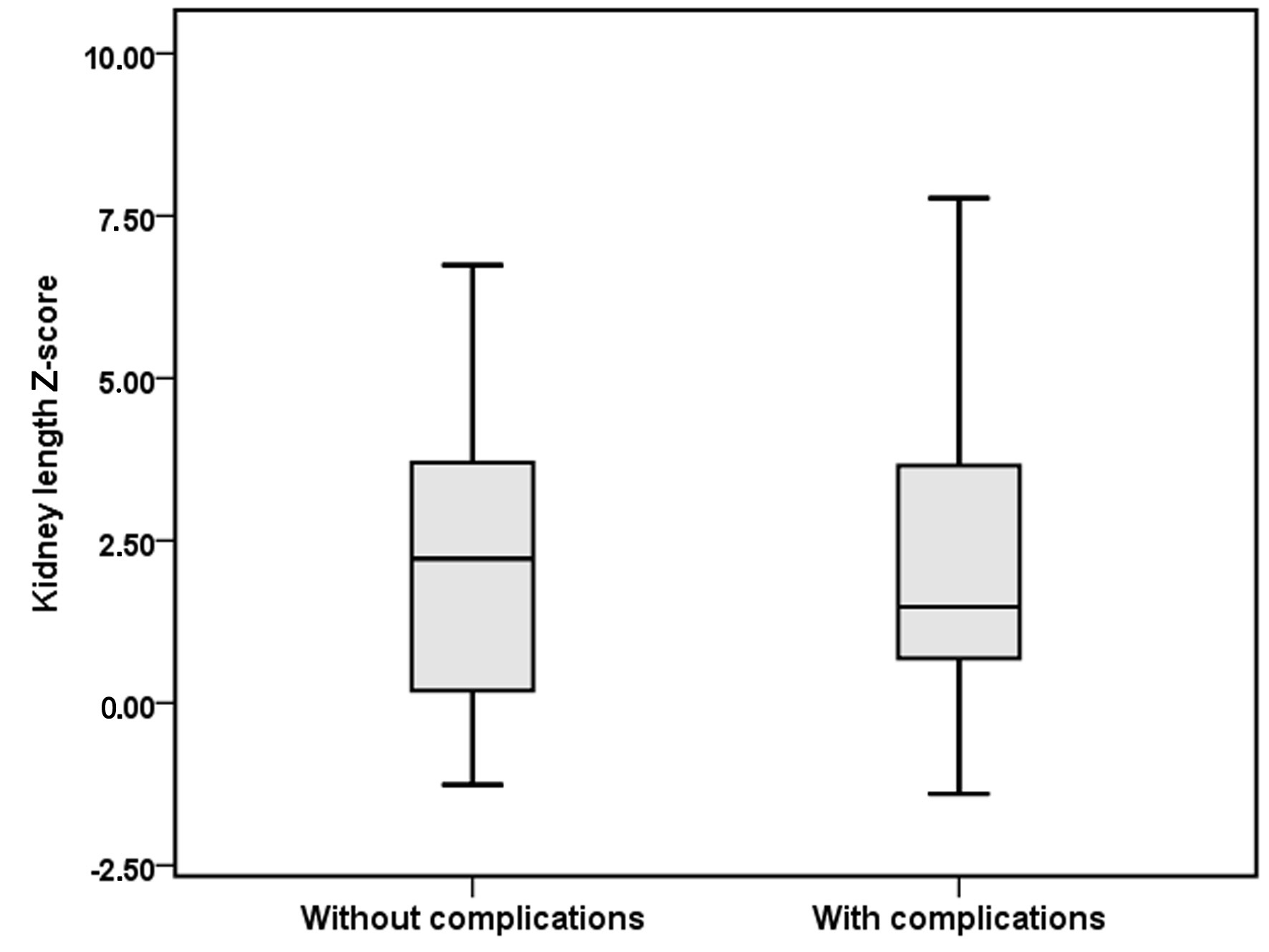 Click for large image | Figure 1. Relationship between kidney length Z-score and the presence of ADPKD-related complications in children with ADPKD. Box-and-whisker plot showing the distribution of kidney length Z-scores in patients with and without complications. ADPKD: autosomal dominant polycystic kidney disease. |
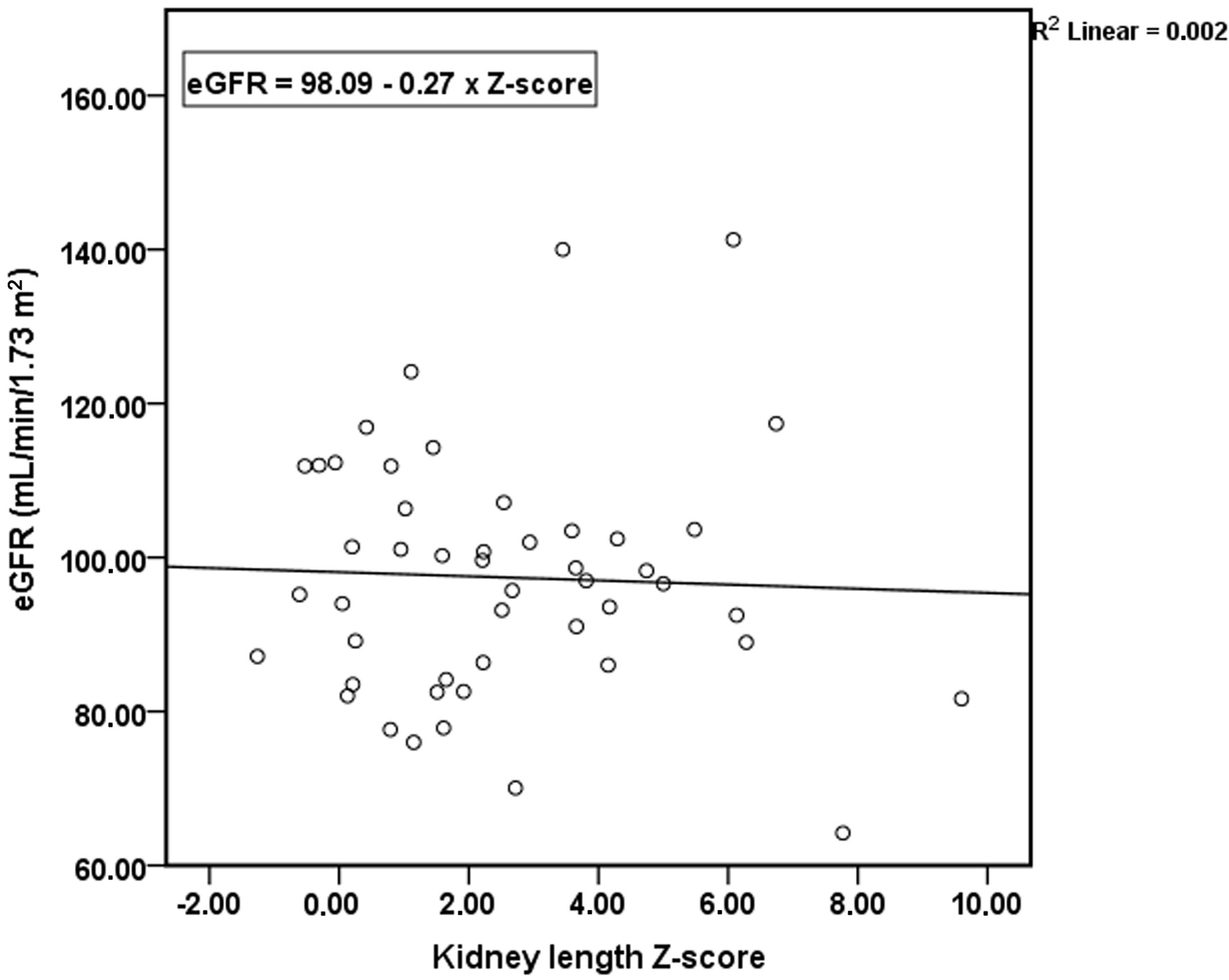 Click for large image | Figure 2. Relationship between kidney length Z-score and estimated glomerular filtration rate (eGFR) in children with ADPKD. Scatter plot illustrating eGFR values across kidney length Z-scores in the study cohort. ADPKD: autosomal dominant polycystic kidney disease. |
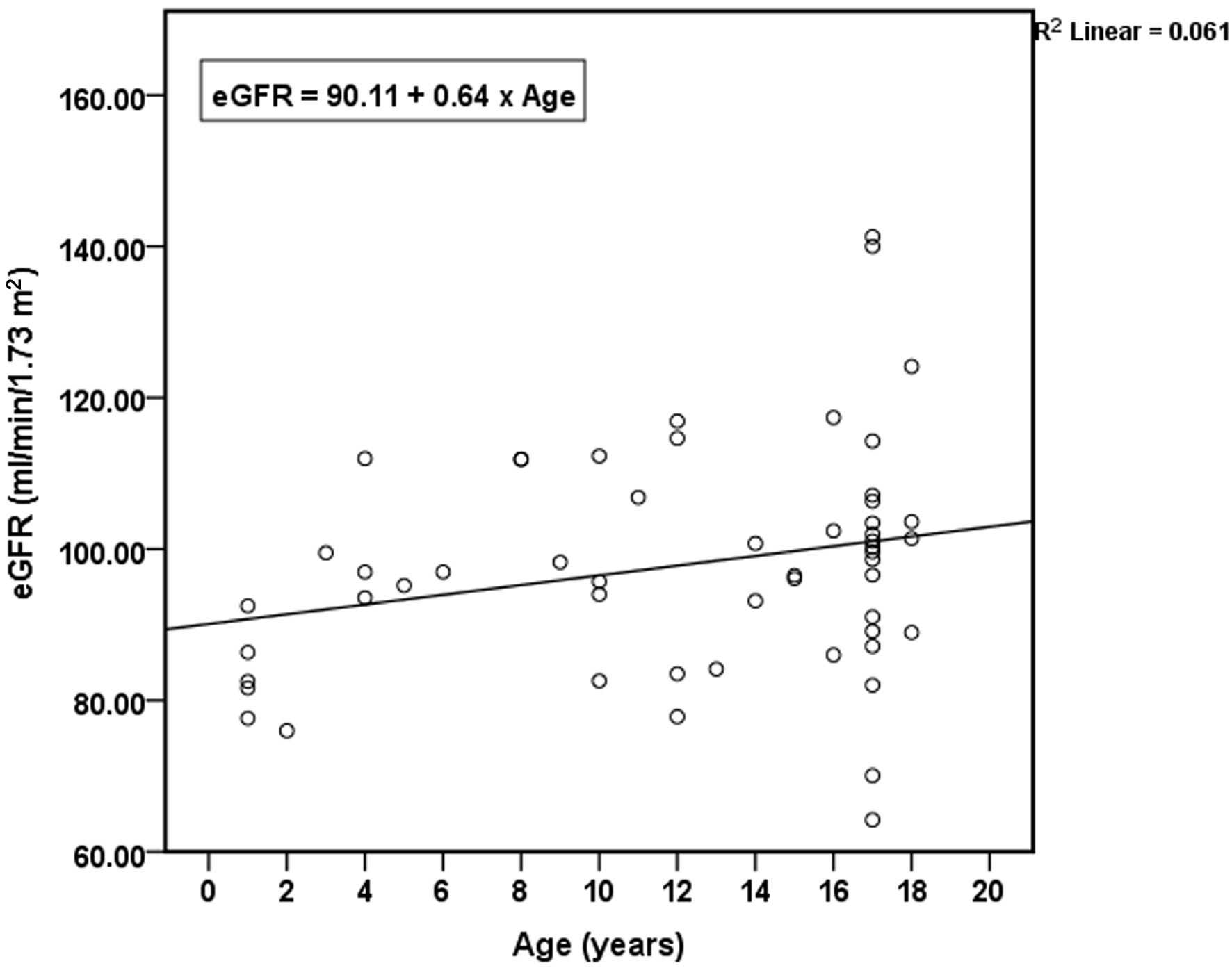 Click for large image | Figure 3. Relationship between age and estimated glomerular filtration rate (eGFR) in children with ADPKD. Scatter plot with linear regression showing the association between age and eGFR. ADPKD: autosomal dominant polycystic kidney disease. |
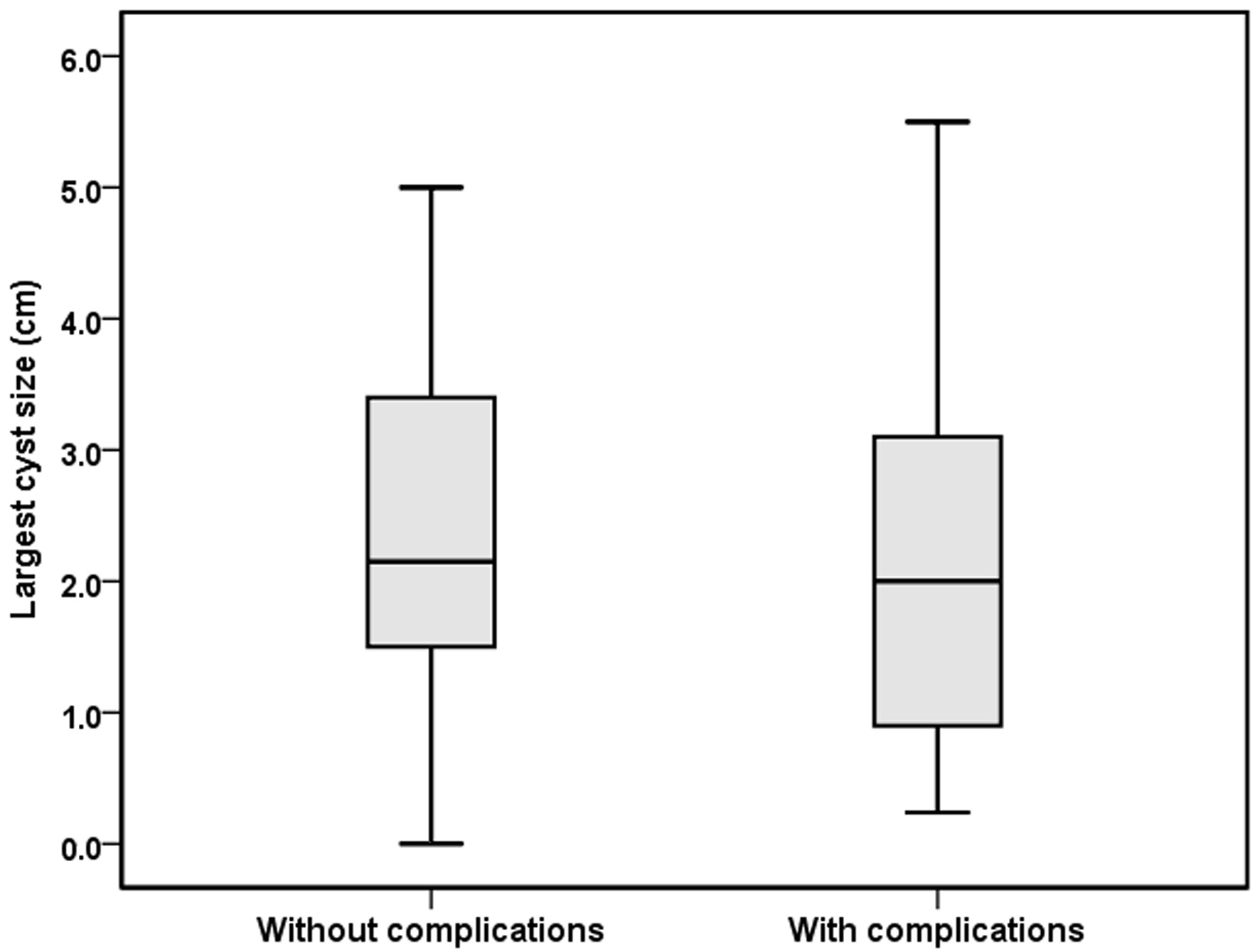 Click for large image | Figure 4. Relationship between largest cyst dimension and the presence of ADPKD-related complications in children with ADPKD. Box-and-whisker plot showing the distribution of largest cyst dimensions in patients with and without complications. ADPKD: autosomal dominant polycystic kidney disease. |
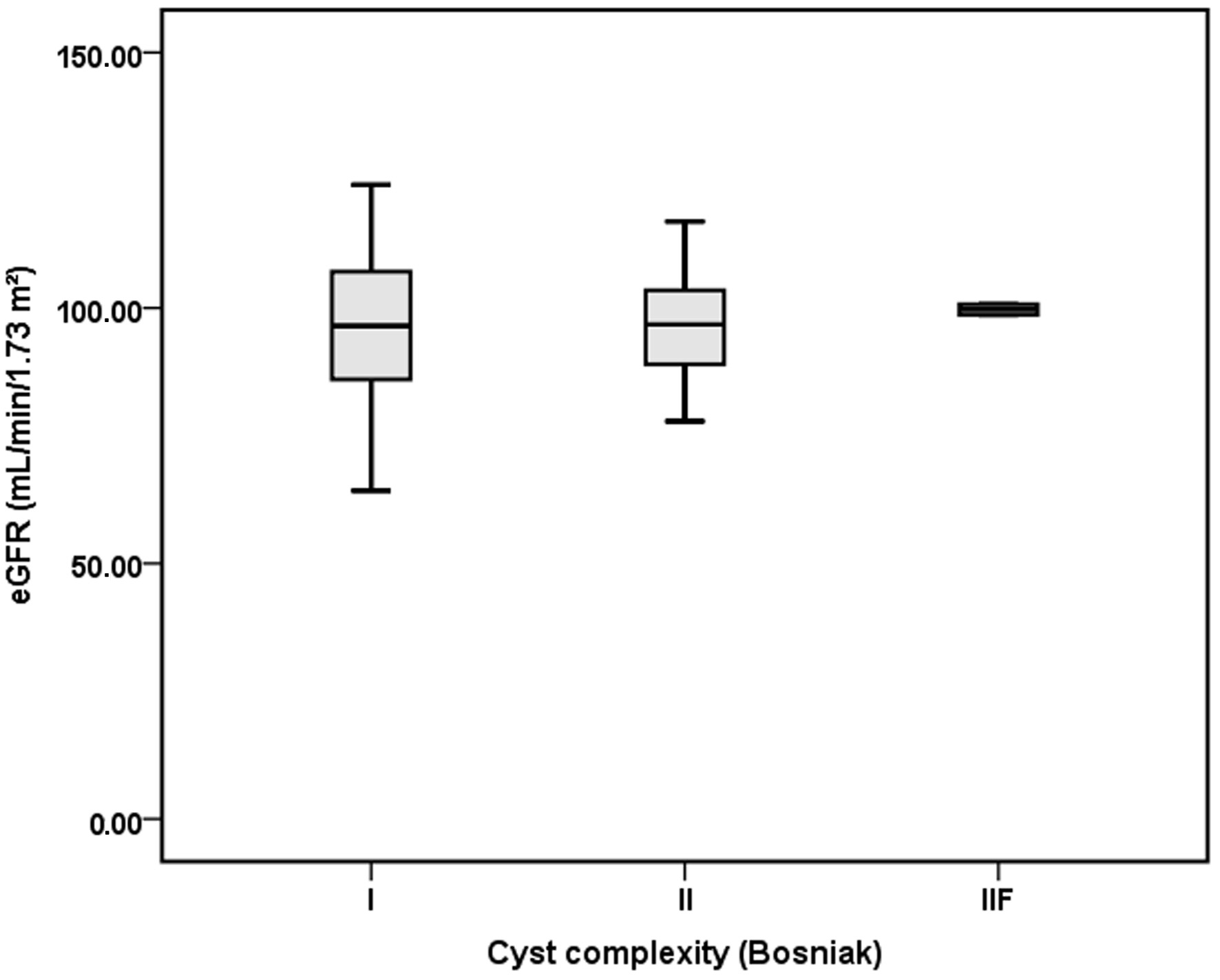 Click for large image | Figure 5. Relationship between cyst complexity and estimated glomerular filtration rate (eGFR) in children with ADPKD. Box-and-whisker plot showing eGFR values according to Bosniak cyst complexity categories (II and IIF). ADPKD: autosomal dominant polycystic kidney disease. |
| Discussion | ▴Top |
Consistent with KDIGO 2025 recommendations and previous pediatric studies, our cohort demonstrates that ADPKD may manifest clinically early in life, with a substantial proportion of affected children developing complications during follow-up. Hypertension and proteinuria, well-established contributors to cardiovascular and renal impairment [4, 9], were among the main complications observed. Early identification and treatment of these modifiable factors remain essential to improve long-term outcomes in pediatric ADPKD [1, 9, 11].
In our cohort, maternal transmission was observed in 61.2% of cases. As ADPKD follows an autosomal dominant inheritance pattern, an approximately equal distribution between maternal and paternal transmission would generally be expected. The slightly higher maternal transmission observed in our cohort likely reflects random variation or cohort-specific referral patterns rather than a true parent-of-origin effect.
Enuresis has a reported prevalence of approximately 5–10% in school-aged children, consistent with epidemiological studies reporting a prevalence of around 7% among children and adolescents [12]. Macroscopic hematuria is uncommon in the general pediatric population, while screening studies report asymptomatic hematuria in approximately 0.5–1% of children [13]. Therefore, although these manifestations are not specific to ADPKD, their occurrence in this context may warrant clinical attention. The prevalence of macroscopic hematuria in our cohort (1.5%) was lower than the 10–14% previously reported [11], which may be partially explained by the younger age of several patients. Older children exhibited a significantly higher frequency of complications, suggesting that clinical burden increases progressively with age despite preserved eGFR. Female patients experienced a higher overall frequency of complications, whereas male patients presented with lower eGFR values. These findings may reflect sex-related differences in early disease trajectories, but could also be influenced by sampling variation or referral patterns. Importantly, we did not identify predictive associations between clinical complications and imaging biomarkers such as kidney size, cyst number, or cyst complexity. This contrasts with one previous study that reported correlations between kidney volume metrics and blood pressure [9]. However, pediatric evidence remains limited by modest sample sizes and the lack of comprehensive longitudinal imaging. KDIGO 2025 guidelines emphasize that no validated prognostic imaging biomarker currently exists for children and that risk stratification in this age group relies primarily on clinical monitoring [1].
The higher frequency of complications observed in females was largely driven by urinary tract infections, which are known to be more prevalent in girls in the general pediatric population. Although females were older at the time of evaluation, no significant association between age and eGFR was identified in regression analysis. Therefore, age alone does not appear to explain the observed sex-related differences in renal function, although the limited sample size may reduce statistical power [11].
Previous studies evaluating predictors of disease progression in ADPKD have produced heterogeneous results. Some clinical characteristics, including male sex, early-onset hypertension, recurrent gross hematuria, and overt proteinuria, have been associated with a more rapid decline in eGFR in certain cohorts. However, evidence remains inconsistent across studies, particularly in pediatric populations in which renal function is generally preserved during the early stages of the disease [1].
Among genetic factors, PKD1 mutations are generally associated with a more severe disease course and a faster decline in eGFR compared with PKD2 mutations. In our cohort, genetic testing was performed in seven patients, all of whom carried PKD1 variants, precluding genotype–phenotype comparisons [1].
Imaging biomarkers such as total kidney volume have also been identified as predictors of renal function decline in adult ADPKD cohorts. However, these parameters were not systematically available in our study, where kidney size assessment relied on ultrasonographic measurements [9].
Finally, previous studies have shown that eGFR decline may be associated with hypertension occurring before the age of 35, as well as with early episodes of hematuria, cyst growth, or infection. The relatively young age of our cohort and the limited duration of follow-up may therefore explain why we did not identify significant predictors of eGFR decline.
The absence of approved disease-modifying therapies for pediatric patients currently restricts management to the prevention and treatment of ADPKD-related complications. Although tolvaptan is approved for adults with rapidly progressive disease and is under investigation in children and adolescents, early recognition of disease manifestations and close longitudinal follow-up remain crucial to optimize supportive care and to ensure timely eligibility for potential future disease-modifying therapies [1, 14]. Accordingly, further studies are needed to identify reliable predictors of complications, refine management strategies, and better characterize the multisystem impact of ADPKD in childhood [6, 14].
Our study is limited by its retrospective design and sample size. Nevertheless, it provides 25 years of real-world data from a pediatric ADPKD cohort, contributing valuable insights into the early clinical course of the disease in childhood.
Conclusions
ADPKD may demonstrate measurable clinical impact during childhood, with relevant rates of urinary tract infections, proteinuria, and hypertension. Although no reliable predictors of complications were identified in our cohort, previous studies have suggested associations between kidney size, cyst number, and hypertension. Larger longitudinal pediatric cohorts are therefore needed to further investigate potential early prognostic markers.
In the absence of validated predictors, early detection and management of modifiable complications, particularly hypertension and proteinuria, remain essential. Both conditions are treatable, and timely intervention may help delay the progression of chronic kidney disease. Structured follow-up, lifestyle counseling, and strict control of blood pressure and proteinuria are key strategies to slow disease progression in children with ADPKD.
Acknowledgments
The authors thank the clinical staff of the Pediatric Nephrology Unit for their contribution to patient care and data collection.
Financial Disclosure
This study did not receive any specific funding.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
According to national legislation and institutional policy, informed consent was not required.
Author Contributions
Mariana Costa and Joao Dias: conceptualization, study design, data analysis, and interpretation, drafting of the manuscript, critical intellectual content, and final approval of the version to be published. Marta Machado, Catarina Neves, Carmen do Carmo, and Carolina Cordinha: Critical revision of the manuscript for important intellectual content and final approval of the version to be published. Clara Gomes: supervision, critical revision of the manuscript for important intellectual content, and final approval of the version to be published. All authors agree to be accountable for all aspects of the work and ensure that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
ADPKD: autosomal dominant polycystic kidney disease; eGFR: estimated glomerular filtration rate; KDIGO: Kidney Disease, Improving Global Outcomes
| References | ▴Top |
- Torres VE, Ahn C, Barten TRM, Brosnahan G, Cadnapaphornchai MA, Chapman AB, Cornec-Le Gall E, et al. KDIGO 2025 clinical practice guideline for the evaluation, management, and treatment of autosomal dominant polycystic kidney disease (ADPKD): executive summary. Kidney Int. 2025;107(2):234-254.
doi pubmed - Cadnapaphornchai MA, Dell KM, Gimpel C, Guay-Woodford LM, Gulati A, Hartung EA, Liebau MC, et al. Polycystic kidney disease in children: the current status and the next horizon. Am J Kidney Dis. 2025;86(3):383-392.
doi pubmed - Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, Paterson AD, et al. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J Am Soc Nephrol. 2018;29(10):2593-2600.
doi pubmed - Seeman T, Pohl M, John U. Proteinuria in children with autosomal dominant polycystic kidney disease. Minerva Pediatr. 2018;70(5):413-417.
doi pubmed - Massella L, Mekahli D, Paripovic D, Prikhodina L, Godefroid N, Niemirska A, Agbas A, et al. Prevalence of hypertension in children with early-stage ADPKD. Clin J Am Soc Nephrol. 2018;13(6):874-883.
doi pubmed - Lai S, Mastroluca D, Matino S, Panebianco V, Vitarelli A, Capotosto L, Turinese I, et al. Early markers of cardiovascular risk in autosomal dominant polycystic kidney disease. Kidney Blood Press Res. 2017;42(6):1290-1302.
doi pubmed - Helal I, Reed B, McFann K, Yan XD, Fick-Brosnahan GM, Cadnapaphornchai M, Schrier RW. Glomerular hyperfiltration and renal progression in children with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6(10):2439-2443.
doi pubmed - Benz EG, Hartung EA. Predictors of progression in autosomal dominant and autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2021;36(9):2639-2658.
doi pubmed - Seeman T, Dusek J, Vondrichova H, Kyncl M, John U, Misselwitz J, Janda J. Ambulatory blood pressure correlates with renal volume and number of renal cysts in children with autosomal dominant polycystic kidney disease. Blood Press Monit. 2003;8(3):107-110.
doi pubmed - Cornec-Le Gall E, Audrezet MP, Rousseau A, Hourmant M, Renaudineau E, Charasse C, Morin MP, et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27(3):942-951.
doi pubmed - Reddy BV, Chapman AB. The spectrum of autosomal dominant polycystic kidney disease in children and adolescents. Pediatr Nephrol. 2017;32(1):31-42.
doi pubmed - Adisu MA, Habtie TE, Munie MA, Bizuayehu MA, Zemariam AB, Derso YA. Global prevalence of nocturnal enuresis and associated factors among children and adolescents: a systematic review and meta-analysis. Child Adolesc Psychiatry Ment Health. 2025;19(1):23.
doi pubmed - Park E, Kim SW, Kim SJ, Baek M, Ahn YH, Yang EM, Cho MH, et al. Asymptomatic hematuria in children: Korean Society of Pediatric Nephrology recommendations for diagnosis and management. Kidney Res Clin Pract. 2024;43(5):565-574.
doi pubmed - Dudley J, Winyard P, Marlais M, Cuthell O, Harris T, Chong J, Sayer J, et al. Clinical practice guideline monitoring children and young people with, or at risk of developing autosomal dominant polycystic kidney disease (ADPKD). BMC Nephrol. 2019;20(1):148.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.