| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://ijcp.elmerpub.com |
Case Report
Volume 000, Number 000, October 2025, pages 000-000

A Case of Homozygous Beta+-Thalassemia
Angela Allena, e ![]() , Abdou Gaia, Yarreh Susoa, Nicholas Parkinb, David Reesc, Stephen J. Allena, d
, Abdou Gaia, Yarreh Susoa, Nicholas Parkinb, David Reesc, Stephen J. Allena, d
aDepartment of Paediatrics, Edward Francis Small Teaching Hospital, Banjul, The Gambia
bMolecular Pathology Laboratory, Synnovis at King’s College Hospital, London, UK
cDepartment of Paediatric Haematology, Kings College London, London, UK
dDepartment of Clinical Sciences, Liverpool School of Tropical Medicine, Pembroke Place, Liverpool, UK
eCorresponding Author: Angela Allen, Department of Paediatrics, Edward Francis Small Teaching Hospital, Banjul, The Gambia
Manuscript submitted July 22, 2025, accepted September 18, 2025, published online October 10, 2025
Short title: A Case of Homozygous β+ Thalassemia
doi: https://doi.org/10.14740/ijcp1018
| Abstract | ▴Top |
Although sickle cell disease is well documented in The Gambia, West Africa, and α-thalassemia has also been identified, we are not aware that β-thalassemia has been reported previously. We presented here a 5-year-old Gambian female with homozygous β+-thalassemia. Genetic analysis revealed the c.316-3C>A variant (also known as IVSII-848(C>A)) in the HBB gene. Since at least one hemoglobin subunit β mutation for β-thalassemia is present in The Gambia, and carrier status of sickle hemoglobin (HbS) is common, the possibility of homozygous β-thalassemia and HbS β-thalassemia should be considered in individuals with the appropriate clinical features and consistent laboratory findings.
Keywords: Thalassemia; Anemia; Splenomegaly; Genetic analysis
| Introduction | ▴Top |
Hemoglobinopathies are a major cause of anemia in sub-Saharan Africa. In The Gambia, the sickle hemoglobin (HbS) carrier frequency is 15-20% [1, 2], but the thalassemias are less well characterized. α-thalassemia has been reported [3, 4], but β-thalassemia has not previously been described, despite its presence in neighboring West African countries [5-9]. We report the first documented case of homozygous β+-thalassemia in The Gambia, caused by the rare c.316-3C>A (IVSII-848 C>A) variant, underscoring the need to consider β-thalassemia in the differential diagnosis of severe anemia in this region.
| Case Report | ▴Top |
A 5-year-old girl was referred to the hematology clinic, Edward Francis Small Teaching Hospital, Banjul, The Gambia, with suspected leukemia. She had a 4-year history of recurrent pallor, tiredness and progressively increasing abdominal distension. There was no history of painful episodes or dactylitis. She had required several hospital admissions and received three blood transfusions.
The pregnancy and delivery were uneventful. Immunizations were reported to be up to date, but the infant welfare card was lost. The parents were in a monogamous, consanguineous marriage and stated Mandinka as their ethnicity. The patient was the second of four children; the first pregnancy ended in a preterm stillbirth; the third child also had a history of abdominal swelling and blood transfusions and died at age 3 years; the fourth child, a 1-year-old girl, was alive and well. The father reported that he had two younger female siblings and three older male siblings, all of whom were alive and well. Other than the consanguineous marriage, no information was available about the mother’s family.
On examination, the patient was markedly pale but hemodynamically stable. There was a prominent forehead, flat nasal bridge, marked prominence of the maxilla, gingival hypertrophy and disordered dentition. There was no bruising, petechiae, jaundice, or lymphadenopathy. The abdomen was grossly distended, soft with non-tender, smooth hepatomegaly of 11 cm below the right costal margin and splenomegaly of 16 cm below the left costal margin (Fig. 1). A review of the other systems was unremarkable. There was severe stunting (height-for-age z score < -3) but no wasting (weight-for-height z score > 1).
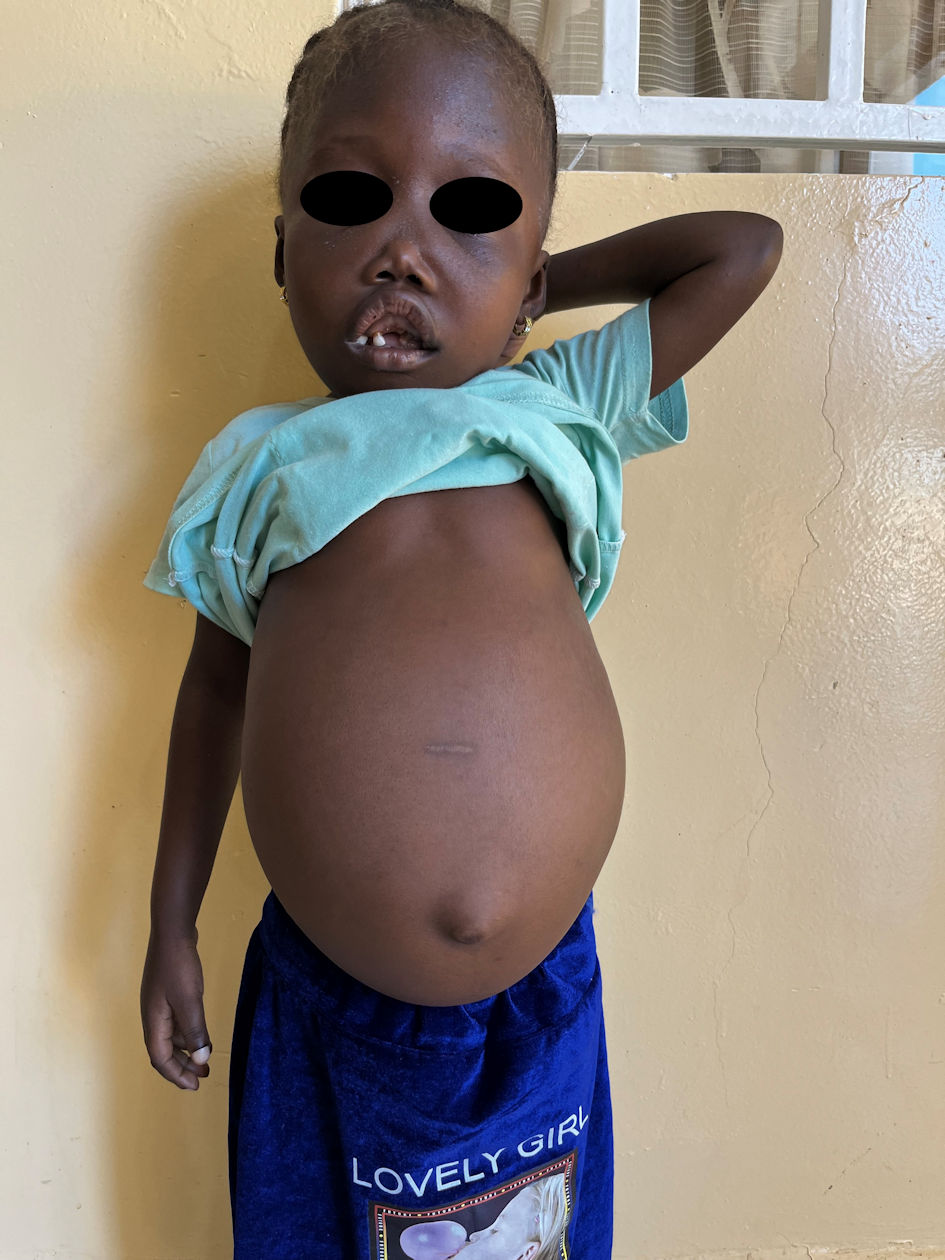 Click for large image | Figure 1. Clinical image. Note the marked pallor, prominent forehead, flat nasal bridge, marked prominence of the maxilla, gingival hypertrophy with disordered dentition and grossly distended abdomen. |
Based on the history and examination, particularly the characteristic facies and massive splenomegaly, a clinical diagnosis of β-thalassemia major was suspected. Abdominopelvic ultrasound examination confirmed hepatosplenomegaly.
Full blood count showed severe microcytic anemia, low red blood cell count and a raised reticulocyte count (Table 1). The automated white blood cell count was markedly raised with lymphocytosis and a reduced proportion of granulocytes (Table 1). Microscopy of a Leishman’s stained thin blood film revealed markedly abnormal red cell morphology, including anisocytosis, hypochromasia, poikilocytosis, numerous nucleated red cells, fragmented cells, target cells and teardrop cells. There was no evidence of marked leukocytosis (Fig. 2). Cellulose acetate hemoglobin (Hb) electrophoresis of a red cell lysate revealed predominantly fetal hemoglobin (HbF) with some adult hemoglobin (HbA) and HbA2.
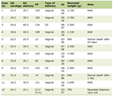 Click to view | Table 1. Hematological Findings |
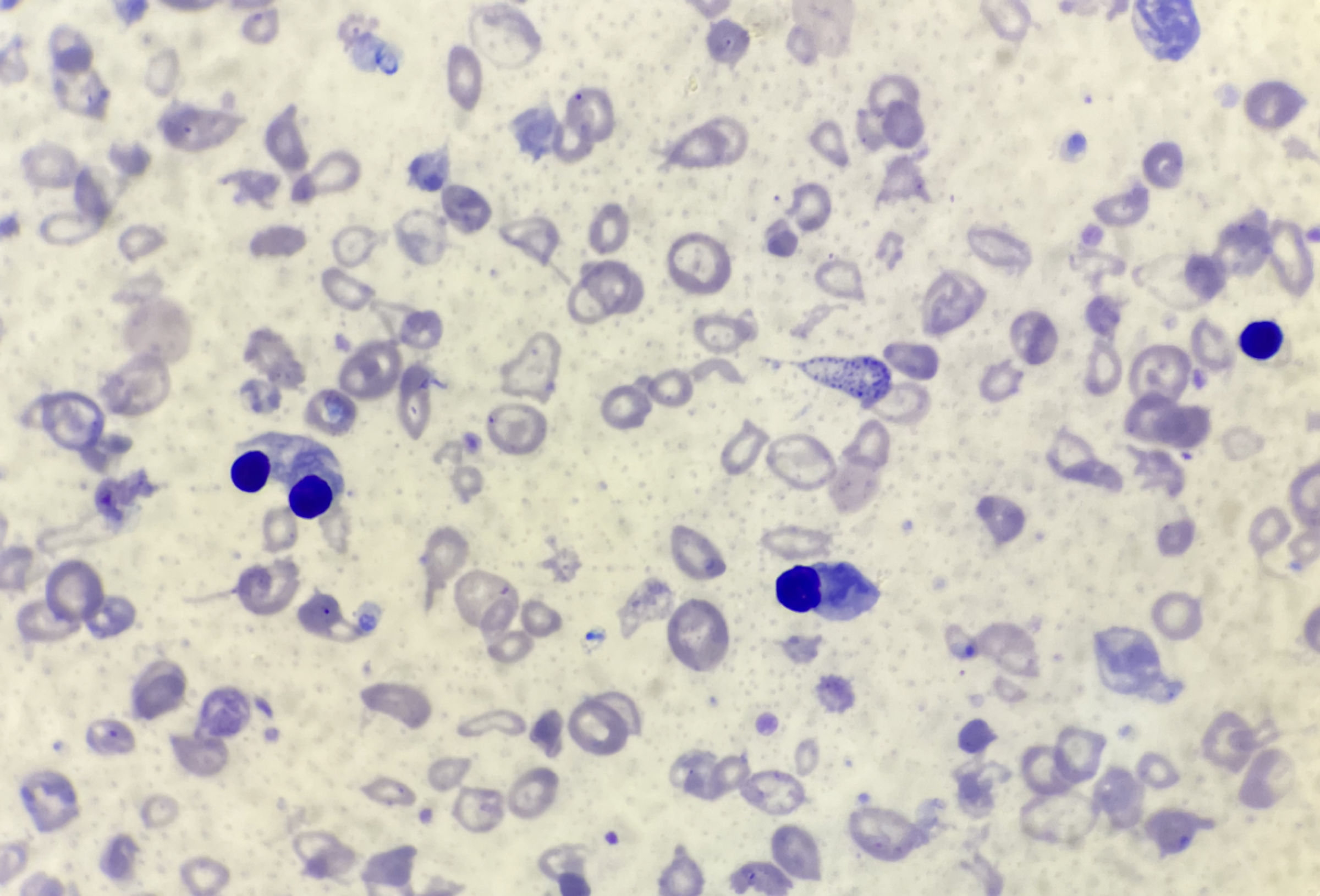 Click for large image | Figure 2. Thin blood film. Thin blood film fixed in methanol and stained with Leishman stain; × 1,000 magnification. Note the markedly abnormal red cell morphology, including anisocytosis, hypochromasia, poikilocytosis, numerous nucleated red cells, fragmented cells, target cells and tear drop cells and no evidence of a marked leukocytosis. |
In the patient’s father, there was mild anemia, microcytosis and raised red blood cell count. Cellulose acetate electrophoresis of the father’s red cell lysate revealed HbA and an elevated HbA2. The proportion of HbA2, quantified by elution and spectrophotometry, was raised. Microscopy of the thin blood film showed microcytosis, poikilocytosis, target cells and some hypochromasia. It was not possible to obtain a blood sample from the mother.
The clinical diagnosis of severe thalassemia intermedia was confirmed by genetic analysis in a molecular pathology diagnostic laboratory, with appropriate quality control. Next-generation sequencing of the Hb subunit β (HBB) gene revealed homozygous β+-thalassemia due to the c.316-3C>A variant (also known as IVSII-848(C>A)). Multiplex ligation-dependent probe amplification (MLPA) analysis did not detect any deletions or duplications of the β-globin gene cluster, nor evidence of delta β-thalassemia, deletional hereditary persistence of fetal hemoglobin (HPFH), or other deletions causing a hemizygous state. In addition, MLPA analysis did not detect any deletions or duplications in the α-globin gene cluster; therefore, there is no evidence of deletional α-thalassemia or additional α-globin genes that may exacerbate the beta thalassemia phenotype.
Testing of the father’s DNA revealed that he is a β+-thalassemia heterozygote and carries the c.316-3C>A variant.
| Discussion | ▴Top |
Although α-thalassemia has been reported in The Gambia [3, 4], and β-thalassemia has been described in other countries in West Africa, including Liberia [5], Nigeria [6], Guinea Bissau [7], Mauritania [8] and neighboring Senegal [9], to the best of our knowledge, this is the first reported case of homozygous β-thalassemia in The Gambia.
The c.316-3C>A (IVSII-848(C>A) variant identified in this child and her father is rare, with a global allele frequency of 0.00002 [10] and has not been described previously in West Africa. It was first described in 1988, in a black patient with β-thalassemia from southern USA [11] and has subsequently been found in Egyptian [12-14], Iranian [12] and Syrian [15] patients, in both homozygous and compound heterozygous β-thalassemia and is associated with a severe phenotype. This rare variant may have arisen de novo in this Gambian family or may be due to intermarriage outside of the region.
A rare mutation comprising of an insertion of thymidine between codons 42 and 43 in exon 2 of the β-globin gene, altering the nucleotide sequence of codon 43 has been reported in a 17-year-old black male with sickle cell disease (SCD, HbS-β0 phenotype) residing in Spain. Although this young man was stated to have been born in The Gambia, his ethnicity is unclear [16].
Recognition that there is at least one β-thalassemia gene present in The Gambia is important for distinguishing homozygous β-thalassemia from SCD because clinical management differs especially with respect to blood transfusion and iron chelation requirements and the role of hydroxyurea. In addition, given the high frequency of the sickle cell gene in The Gambia, previously estimated to be 19.5% [1] and 15.9% [2], this case highlights the likelihood of HbS/β-thalassemia in patients attending hematology clinics. This is especially important where diagnosis is based only on point-of-care, rapid diagnostic tests such as the HemoType SC (Silverlake Research Corporation, California, USA). Although this test identifies HbS, C and A, additional tests applicable to low-resource settings, such as Hb electrophoresis and elution, are required to identify raised HbA2 consistent with β-thalassemia trait, as well as raised HbF and reduced HbA in β+-thalassemia homozygotes. In addition, the referral diagnosis of suspected leukemia was based on the clinical presentation and an apparently markedly raised white cell count. However, many automated hematology analyzers miscount nucleated red cells as white blood cells [17], and this can be resolved by examination of a thin blood film, as in this case.
As in this case, homozygous β-thalassemia can be differentiated from sickle cell anemia (SCA), the most common hemoglobinopathy in this region, based on characteristic facial features resulting from a greater degree of extramedullary hematopoiesis, such as marked gingival hypertrophy and disordered dentition. In addition, dactylitis is common in SCA but does not occur in homozygous β-thalassemia. In contrast, massive splenomegaly is more common in the thalassemias than SCA. The diagnosis can also be supported by a consistent family history, such as another child with a likely inherited anemia and a similar clinical picture as occurred in this family. Finally, as in the father of this case, compatible hematological findings - including low red cell indices and a raised HbA2 in first-degree relatives - support the diagnosis.
HbS/β+ thalassemia can be suspected in patients with HbAS on a rapid diagnostic test who present with more severe clinical manifestations, such as bone pain and blood transfusion requirement [18], than usually occur in sickle cell trait. It should also be noted that in cases of HbS/β0 thalassemia, a rapid diagnostic test such as HemoType SC would only detect HbS due to the absence of HbA in this condition [19].
The implications for newborn screening are complex. At the time of writing, neonatal screening tests utilized in The Gambia are limited to the detection of HbA, S and C (HemoType SC test) and are unable to detect infants born with β0-thalassemia or differentiate between HbSS and HbS/β0-thalassemia. Furthermore, β+-homozygotes, as in our case, would likely be identified as normal due to the presence of HbA and the absence of abnormal Hbs S and C. This complexity highlights that, in addition to newborn screening currently available in The Gambia, additional tests are required in infants and children with equivocal test results or clinical features consistent with a hemoglobinopathy including genetic analyses, Hb electrophoresis later in infancy and testing of parents.
Limitations of our report are that we were unable to obtain and test a blood sample from the mother, to get clear details of the family pedigree and undertake screening of the other family members due to their remote location. A more detailed family pedigree may have helped differentiate between a de-novo mutation and gene flow from other populations. Furthermore, the remote location also compromised clinical follow-up of the child.
This case highlights that combining clinical and laboratory expertise is required for the reliable diagnosis of the hemoglobinopathies and is achievable even in limited resource settings.
Acknowledgments
We would like to thank the child and her family.
Financial Disclosure
This research did not receive any external financial support.
Conflict of Interest
The authors report that there is no conflict of interest to declare.
Informed Consent
Written informed consent was obtained from the father of the child to publish the information and images in this case report.
Author Contributions
AA and SA conceived and designed the analysis. AG and YS collected the data. NP, DR, and AA contributed to the data and analysis tools. NP, DR, and AA performed the analysis. AA, SA, and DR wrote the paper.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
Hb: hemoglobin; HbA: adult hemoglobin; HBB: hemoglobin subunit β; SCA: sickle cell anemia
| References | ▴Top |
- Allen SJ, Bennett S, Riley EM, Rowe PA, Jakobsen PH, O'Donnell A, Greenwood BM. Morbidity from malaria and immune responses to defined Plasmodium falciparum antigens in children with sickle cell trait in The Gambia. Trans R Soc Trop Med Hyg. 1992;86(5):494-498.
doi pubmed - Cox SE, Doherty CP, Atkinson SH, Nweneka CV, Fulford AJ, Sirugo G, Rockett KA, et al. Haptoglobin genotype, anaemia and malaria in Gambian children. Trop Med Int Health. 2008;13(1):76-82.
doi pubmed - Abdalla SH, Corrah PT, Higgs DR. Alpha-thalassaemia in the Gambia. Trans R Soc Trop Med Hyg. 1989;83(3):420.
doi pubmed - Allen SJ, Rowe P, Allsopp CE, Riley EM, Jakobsen PH, Hill AV, Greenwood BM. A prospective study of the influence of alpha thalassaemia on morbidity from malaria and immune responses to defined Plasmodium falciparum antigens in Gambian children. Trans R Soc Trop Med Hyg. 1993;87(3):282-285.
doi pubmed - Willcox MC, Beckman L. Haemoglobin variants, beta-thalassaemia and G-6-PD types in Liberia. Hum Hered. 1981;31(6):339-347.
doi pubmed - Adeyemo T, Ojewunmi O, Oyetunji A. Evaluation of high performance liquid chromatography (HPLC) pattern and prevalence of beta-thalassaemia trait among sickle cell disease patients in Lagos, Nigeria. Pan Afr Med J. 2014;18:71.
doi pubmed - Masmas TN, Garly ML, Lisse IM, Rodriques A, Petersen PT, Birgens H. Inherited hemoglobin disorders in Guinea-Bissau, West Africa: a population study. Hemoglobin. 2006;30(3):355-364.
doi pubmed - Mahmoud T, Sahli C, Hadj Fredj S, Amri Y, Othmani R, Mohamed GS, Zein E, et al. Epidemiological and molecular study of hemoglobinopathies in Mauritanian patients. Mol Genet Genomic Med. 2022;10(10):e2048.
doi pubmed - Gueye Tall F, Martin C, Malick Ndour EH, Deme Ly I, Renoux C, Chillotti L, Veyrenche N, et al. Genetic background of the sickle cell disease pediatric population of Dakar, Senegal, and characterization of a novel frameshift beta-thalassemia mutation [HBB: c.265_266del; p.Leu89Glufs*2]. Hemoglobin. 2017;41(2):89-95.
doi pubmed - VCV000015451.113-ClinVar-NCBI. Available at ncbi.nlm.nih.gov. Accessed November 11, 2024.
- Gonzalez-Redondo JM, Stoming TA, Lanclos KD, Gu YC, Kutlar A, Kutlar F, Nakatsuji T, et al. Clinical and genetic heterogeneity in black patients with homozygous beta-thalassemia from the southeastern United States. Blood. 1988;72(3):1007-1014.
pubmed - Wong C, Antonarakis SE, Goff SC, Orkin SH, Forget BG, Nathan DG, Giardina PJ, et al. Beta-thalassemia due to two novel nucleotide substitutions in consensus acceptor splice sequences of the beta-globin gene. Blood. 1989;73(4):914-918.
pubmed - Elmezayen AD, Kotb SM, Sadek NA, Abdalla EM. beta-Globin mutations in Egyptian patients with beta-thalassemia. Lab Med. 2015;46(1):8-13.
doi pubmed - Elhalfawy KH, Daif A, Shaalan O. Detection of common β thalassemia mutations among Egyptian patients. Egypt J Genet Cytol. 2017;46(1):111-119.
- Shoujaa A, Mukhalalaty Y, Murad H, Al-Quobaili F. Description of a rare beta-globin gene mutation, IVS-II-848 (C>A) (HBB: c.316-3C>A) in association with IVS-I-1 (G>A) (HBB: c.92 + 1G>A), observed in a Syrian family: a case report. Hemoglobin. 2019;43(4-5):283-285.
doi pubmed - Marino SFP, Gradilla PR, Gonzalez Fernandez FA, Martinez AMV, Martinez SM, Bartol Puyal FA, Palomar EV, et al. Sickle cell disease associated with thalassemia; description of a rare mutation. Clin Biochem. 2021;94:80-82.
doi pubmed - Gulati G, Uppal G, Gong J. Unreliable automated complete blood count results: causes, recognition, and resolution. Ann Lab Med. 2022;42(5):515-530.
doi pubmed - Darshana T, Bandara D, Nawarathne U, de Silva U, Costa Y, Pushpakumara K, Pathirage S, et al. Sickle cell disease in Sri Lanka: clinical and molecular basis and the unanswered questions about disease severity. Orphanet J Rare Dis. 2020;15(1):177.
doi pubmed - Adegoke SA, Oladimeji OI, Akinlosotu MA, Akinwumi AI, Matthew KA. HemoTypeSC point-of-care testing shows high sensitivity with alkaline cellulose acetate hemoglobin electrophoresis for screening hemoglobin SS and SC genotypes. Hematol Transfus Cell Ther. 2022;44(3):341-345.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.