| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://ijcp.elmerpub.com |
Case Report
Volume 15, Number 1, March 2026, pages 31-36

Urine Spot Beta2-Microglobulin as a Screening Tool for Dent Disease Type 1
Rehna Kakkidi Rahmana, d ![]() , Farah Alsheikh Bakroua, Ayman El-Hattabb, Divya Pachatc
, Farah Alsheikh Bakroua, Ayman El-Hattabb, Divya Pachatc
aDepartment of Pediatrics, Burjeel Medical City, Abu Dhabi, UAE
bGenetics and Rare Disease Center, Burjeel Medical City, Abu Dhabi, UAE
cDepartment of Genetics, Aster MIMS Hospital, Calicut, Kerala, India
dCorresponding Author: Rehna Kakkidi Rahman, Department of Pediatrics, Burjeel Medical City, Mohamed Bin Zayed City, PO BOX-92510, Abu Dhabi, UAE
Manuscript submitted January 14, 2026, accepted March 14, 2026, published online March 26, 2026
Short title: Urine Beta2 Microglobulin in DDT1
doi: https://doi.org/10.14740/ijcp1042
| Abstract | ▴Top |
Dent disease type 1 (DD1) is a rare X-linked proximal tubulopathy caused by pathogenic variants in CLCN5 gene. It typically presents in childhood with low-molecular-weight proteinuria, hypercalciuria, and variable progression to chronic kidney disease. Early diagnosis is often missed because routine urine dipstick testing detects only albumin and underestimates tubular protein losses. We describe two boys who presented with isolated proteinuria, in whom a marked discrepancy between minimal dipstick albuminuria and elevated 24-h urinary protein excretion raised suspicion for tubular proteinuria. Urine β2-microglobulin levels were markedly increased in both children, supporting proximal tubular dysfunction. Hypercalciuria was identified despite normal serum studies and normal renal ultrasonography. Genetic testing confirmed CLCN5 variants consistent with DD1. The first patient had a de novo large deletion involving exons 3–15, and maternal testing confirmed the absence of carrier status. The second patient carried a familial 3-bp insertion in exon 10 that was reclassified as pathogenic following segregation analysis, with his mother being identified as a heterozygous carrier. Both children were started on angiotensin converting enzyme inhibitors for management of proteinuria. These cases highlight the importance of recognizing tubular proteinuria in children with unexplained proteinuria. Measurement of urine β2-microglobulin is a simple, inexpensive, and highly sensitive tool that can prompt timely evaluation for DD1. Early integration of biochemical markers with targeted genetic testing is essential for accurate diagnosis and appropriate management of this rare but clinically significant condition.
Keywords: Proteinuria; Urine beta2 microglobulin; Dent disease type 1
| Introduction | ▴Top |
Dent disease type 1 (DD1) is a rare X-linked recessive proximal renal tubular disorder characterized by low-molecular-weight proteinuria (LMWP), hypercalciuria, nephrocalcinosis, nephrolithiasis, and progressive chronic kidney disease (CKD) [1]. Approximately 60–70% of cases are caused by pathogenic variants in the CLCN5 gene (DD1), which encodes the ClC-5 chloride/proton exchanger essential for receptor-mediated endocytosis in proximal tubular cells [2, 3]. The remaining cases are predominantly due to OCRL gene variants (Dent disease type 2), which may be associated with syndromic manifestations such as developmental delay, hypotonia, and cataracts.
Although Fanconi syndrome has been described in DD1, children often present with nonspecific features such as isolated proteinuria in the early stages. Importantly, albumin dipstick testing may underestimate the severity of tubular protein loss because low molecular weight proteins (β2-microglobulin, α1-microglobulin, retinol-binding protein) are not detected reliably by dipsticks [4]. Consequently, DD1 is frequently underdiagnosed or misdiagnosed as glomerular disease, and many children undergo unnecessary renal biopsy.
Urine β2-microglobulin is typically markedly elevated in DD1 and serves as a useful initial screening tool, particularly when there is a discrepancy between dipstick albuminuria and total protein excretion. The two cases presented here highlight important and increasingly recognized aspects of DD1, particularly the role of LMWP screening, and the importance of genetic testing even in the absence of classical features or a family history.
| Case Reports | ▴Top |
Case 1
Clinical description
A 3-year-old Indian boy was evaluated following incidental detection of 2+ albuminuria on urine dipstick testing during an intercurrent illness. He was the first child of non-consanguineous parents. Prenatal and postnatal histories were unremarkable, and he was born at term with a birth weight of 3 kg. There was no history of edema, frothy urine, hematuria, recurrent fever, rash, oral ulcers, joint swelling, recurrent urinary tract infections, jaundice, polyuria, polydipsia, bony deformities, pallor, or chronic medication intake. Family history was negative for renal disease.
On examination, he was hemodynamically stable and normotensive. Anthropometric measurements showed weight at the 25th percentile (14 kg) and height at the 35th percentile (95 cm). No edema was present, and systemic examination was unremarkable.
Diagnosis and management
Initial laboratory evaluation (Table 1) revealed normal renal function, electrolytes, hemogram, and liver function tests. Serum albumin and cholesterol were within normal limits. Urinalysis showed 2+ albuminuria with 0–2 white blood cells (WBCs)/high-power field (HPF), 0–1 red blood cell (RBC)/HPF, and no glucosuria. The urine protein-to-creatinine ratio (PCR) was 3.2 mg/mg. Repeat first-morning urine analysis showed similar results, ruling out orthostatic proteinuria. Complement levels (C3, C4), anti-nuclear antibody (ANA), anti-neutrophilic cytoplasmic antibody (ANCA), anti-glomerular basement membrane (GBM) antibodies, and viral serologies (human immunodeficiency virus (HIV), hepatitis B surface antigen (HBsAg), hepatitis C virus (HCV)) were negative. Renal ultrasonography was normal.
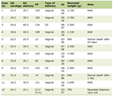 Click to view | Table 1. Laboratory Investigations |
A 24-h urine protein excretion was 42 mg/m2/h (total 605 mg/24 h). The discrepancy between 24-h proteinuria and albumin dipstick findings suggested tubular proteinuria. Spot urinary β2-microglobulin was markedly elevated (> 20,000 ng/mL). Evaluation for tubular dysfunction showed normal venous blood gas, serum calcium, phosphorus, and alkaline phosphatase levels. Urine pH was 5, with no glucosuria. The urine calcium-to-creatinine ratio was elevated (0.65 mg/mg), which was confirmed by 24-h urinary calcium excretion of 5 mg/kg/day.
Based on the presence of LMWP and hypercalciuria without other features of Fanconi syndrome, DD1 was suspected. Although DD1 is inherited in an X-linked recessive manner, there was no family history of renal disease. The mother’s urine β2-microglobulin measurement was normal. Clinical exome sequencing (CES) identified a 166.92-kb contiguous deletion involving exons 3–15 of the CLCN5 gene, classified as “likely pathogenic” according to American College of Medical Genetics (ACMG) criteria, confirming DD1. Maternal testing for the same deletion was negative, indicating a de novo mutation, consistent with her normal urinary β2-microglobulin level.
The child was started on an angiotensin converting enzyme (ACE) inhibitor for proteinuria. At follow-up, urine PCR improved slightly to 2.9 mg/mg. Hydrochlorothiazide was not initiated for hypercalciuria because of the absence of nephrolithiasis or nephrocalcinosis and the higher risk of adverse effects at this young age. Adequate hydration and urinary alkalinization with citrate was advised.
Case 2
Clinical description
A 6-year-old Indian boy presented with a 1-year history of intermittently frothy urine. He was the first child of non-consanguineous parents and was born at term with a birth weight of 3.2 kg after an uncomplicated perinatal period. There was no history of edema, hematuria, rash, jaundice, polyuria, polydipsia, recurrent urinary tract infections, hearing, or visual problems.
A notable family history included renal transplantation in the maternal grandfather, although the cause of kidney failure was unknown. He reportedly had no hearing or visual deficits. On examination, the child was normotensive and well grown (weight 18 kg; height 106 cm). There was no edema, and systemic examination was normal.
Diagnosis and management
Laboratory evaluation (Table 1) revealed normal renal function, hemogram, liver function tests, serum albumin, and cholesterol. First-morning urinalysis showed 2+ albuminuria with 0–2 WBCs/HPF, 0–2 RBCs/HPF, and no glucosuria. The urine PCR was 2.8 mg/mg. Complement levels, ANA, ANCA, anti-GBM antibodies, and viral serologies were normal. Renal ultrasonography was also normal.
A 24-h urine protein excretion was 49 mg/m2/h (820 mg/24 h). Because of the maternal-line family history of renal disease, X-linked Alport syndrome was initially considered; however, there was no hematuria, and ophthalmologic and audiologic evaluations were normal. The mother’s urine dipstick was negative.
Given the X-linked inheritance pattern shared by DD1, tubular protein assessment was performed. The child’s spot urine β2-microglobulin level was markedly elevated (> 20,000 ng/mL), and the mother had borderline elevation (1,460 ng/mL). Venous blood gas and serum electrolytes, calcium, and phosphorus were normal. The 24-h urinary calcium excretion was elevated at 5.9 mg/kg/day, confirming hypercalciuria.
CES identified a hemizygous 3-bp insertion in exon 10 of the CLCN5 gene, initially classified as a variant of uncertain significance (VUS). The same variant was identified in hemizygous form in the maternal grandfather and in heterozygous form in the mother. Segregation analysis supported reclassification of this variant as pathogenic, confirming DD1.
The child was started on an ACE inhibitor for proteinuria. At follow-up, the urine PCR improved to 2.4 mg/mg. Six months later, renal ultrasonography revealed medullary nephrocalcinosis, prompting initiation of urinary alkalinization with citrate.
Genetic reports and urine beta2 microglobulin values are compared in Table 2.
Click to view | Table 2. Urine Beta2 Microglobulin and Genetic Analysis of Cases and Family Members |
| Discussion | ▴Top |
Dent disease is an X-linked proximal tubulopathy that usually presents in childhood but may be overlooked because of the subtle early manifestations. Both children in this series presented with isolated nephrotic range proteinuria, normal renal function, and absence of edema—findings that often lead clinicians to suspect glomerular pathology or dismiss the abnormalities as transient. DD1 results from pathogenic CLCN5 variants on Xp11.23, whereas mutations in OCRL lead to type 2 disease, often with extra-renal features. LMWP is the only manifestation which is present in 100% of cases. Table 3 depicts the frequencies of various manifestations in DD1 [5–11]. Interestingly renal tubular acidosis is exceedingly rare [5].
Click to view | Table 3. Frequency of Clinical Manifestations in DD1 [5–11] |
LMWP is typically 100–1,000 times the upper limit of normal and may be the only abnormality in the initial years of life. Carrier females frequently exhibit mild LMWP, as demonstrated in case 2. A recent large multicenter European cohort study found that although LMWP was universal, only 44% had hypercalciuria and fewer than 70% had nephrocalcinosis at diagnosis, underscoring the evolving nature of the phenotype [12]. This aligns with our cases where nephrocalcinosis was initially absent and developed later in one child.
In both boys, the discrepancy between dipstick albumin and high total protein excretion raised suspicion of a tubular cause. Markedly elevated urine β2-microglobulin confirmed proximal tubular dysfunction. This pattern is increasingly recognized as a diagnostic signature of Dent disease, and several recent reports stress the importance of testing for LMW proteins in boys with unexplained, non-nephrotic proteinuria, especially when hypercalciuria is present [8, 13].
Hypercalciuria was observed in both cases; nephrocalcinosis developed later in case 2. Multifactorial mechanisms have been proposed, including increased calcium delivery to the distal nephron, impaired tubular reabsorption, increased luminal parathyroid hormone (PTH), and elevated 1,25-dihydroxyvitamin D levels [1]. These mechanisms remain incompletely understood. Early identification of hypercalciuria is essential because nephrocalcinosis contributes to CKD progression.
Although LMWP and hypercalciuria strongly suggest Dent disease, genetic confirmation is recommended in all suspected cases because CLCN5 mutations account only for ∼60% and OCRL mutations for ∼15%. Nearly 300 different mutations are described, including missense, nonsense, insertions, deletions, and large copy-number variations [1, 5, 7]. Genotype–phenotype correlations are limited, although mutations affecting the ClC-5 pore or CBS domains may be associated with faster decline in renal function [6, 14, 15].
In case 1, a de novo large deletion (exons 3–15) confirmed the diagnosis and explained the negative maternal screening. In case 2, a complex insertion variant required segregation analysis, which reclassified it from “VUS” to “pathogenic”, highlighting the importance of family evaluation in X-linked disorders.
Management options are limited and mainly supportive measures like ACE inhibitors/angiotensin receptor blockers (ARBs) to reduce proteinuria, thiazides for hypercalciuria when nephrocalcinosis is evident and hydration/urinary alkalinization to prevent stone formation. Emerging research using chemical chaperones (e.g., 4-phenylbutyrate) shows promise in improving ClC-5 processing in specific mutations, suggesting a potential future disease-modifying therapy [16].
The recently published clinical practice recommendations suggest to screen for LMWP/DD1 in any boy presenting with isolated proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, rickets, and CKD of unknown etiology [5]. Figure 1 demonstrates a flow diagram for evaluation in such patients (adapted from Ref. [5]).
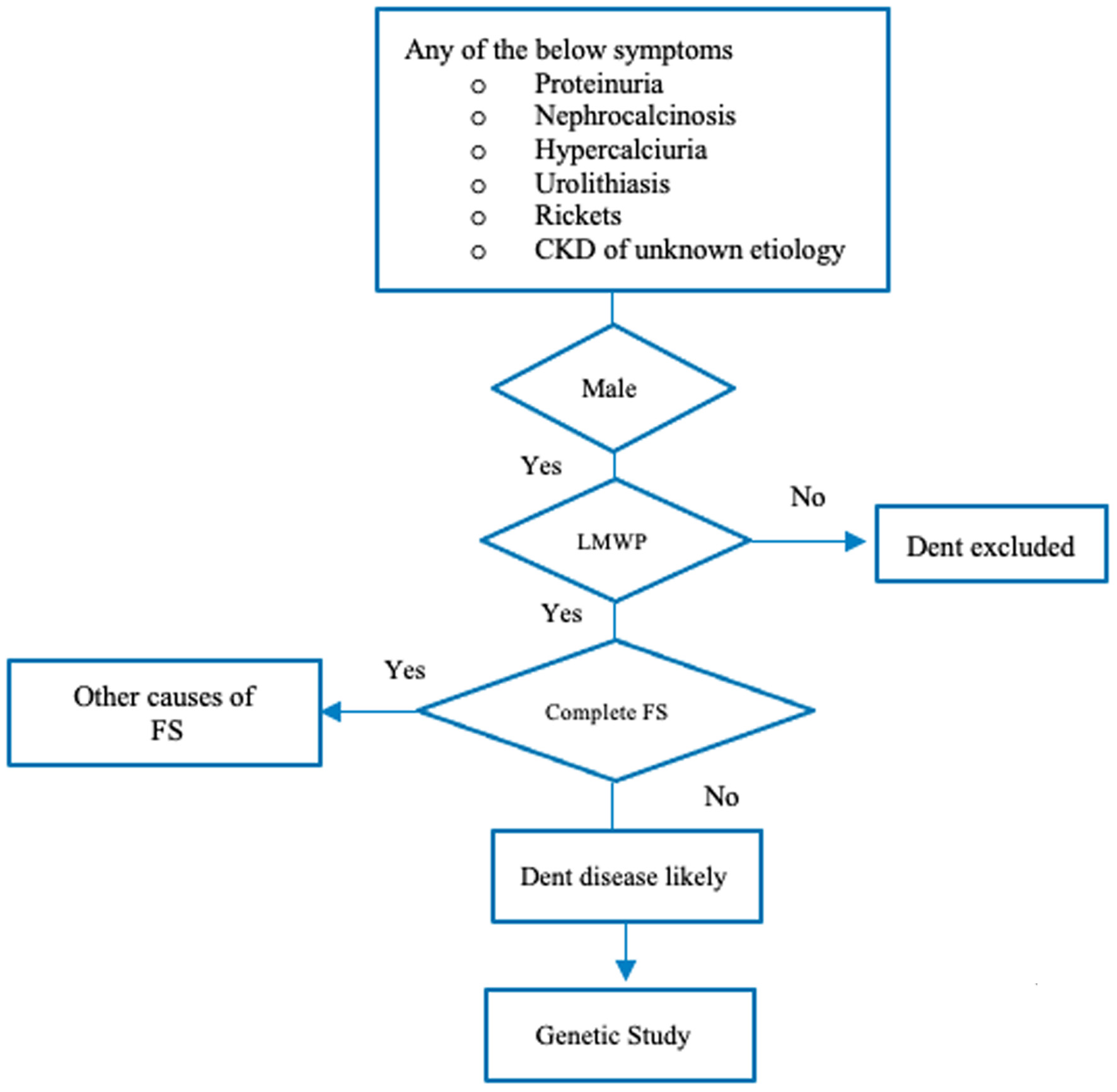 Click for large image | Figure 1. Flow diagram for evaluation of Dent disease (adapted from Reference [5]). CKD: chronic kidney disease; FS: Fanconi syndrome; LMWP: low-molecular-weight proteinuria. |
Conclusions
Dent disease should be considered in any child presenting with isolated or unexplained proteinuria, particularly when: there is a mismatch between dipstick albumin and total protein excretion, features of proximal tubular dysfunction are present, or there is a suggestive family history.
Urine β2-microglobulin serves as a simple and reliable initial screening tool. Genetic testing confirms the diagnosis, enables early monitoring for CKD progression, and facilitates family counseling. Early recognition is essential for timely intervention and optimal long-term outcomes.
Acknowledgments
We acknowledge the parents who had been patient with us during the diagnostic process.
Financial Disclosure
None to declare.
Conflict of Interest
The authors have indicated that they have no conflicts of interest relevant to this article.
Informed Consent
Written informed consent was obtained from both patient’s parents for publication of case report.
Author Contributions
Dr Farah Alsheikh Bakrou (pediatrician) and Dr Rehna Kakkidi Rahman (pediatric nephrologist) worked up the patient and conceptualized the case report, collected details, and prepared the manuscript and also reviewed the manuscript. Dr Ayman El-Hattab and Divya Pachat (geneticist) were involved in interpreting the genetic tests, editing the manuscript and final corrections. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request (genetic study reports).
Abbreviations
ACE: angiotensin converting enzyme; ACMG: American College of Medical Genetics; ARB: angiotensin receptor blocker; ANA: anti-nuclear antibody; ANCA: anti-neutrophilic cytoplasmic antibody; anti-GBM: anti-glomerular basement membrane; CES: clinical exome sequencing; CKD: chronic kidney disease; DD1: Dent disease type 1; HPF: high-power field; HIV: human immunodeficiency virus; HBSAg: hepatitis B surface antigen; HCV: hepatitis C virus; PCR: protein-to-creatinine ratio; PTH: parathyroid hormone; RBC: red blood cell; SS: Sanger sequencing; VUS: variant of uncertain significance; WBC: white blood cell
| References | ▴Top |
- Claverie-Martin F, Ramos-Trujillo E, Garcia-Nieto V. Dent's disease: clinical features and molecular basis. Pediatr Nephrol. 2011;26(5):693-704.
doi pubmed - Lloyd SE, Gunther W, Pearce SH, Thomson A, Bianchi ML, Bosio M, Craig IW, et al. Characterisation of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders. Hum Mol Genet. 1997;6(8):1233-1239.
doi pubmed - Devuyst O, Thakker RV. Dent's disease. Orphanet J Rare Dis. 2010;5:28.
doi pubmed - Blanchard A, Curis E, Guyon-Roger T, Kahila D, Treard C, Baudouin V, Berard E, et al. Observations of a large Dent disease cohort. Kidney Int. 2016;90(2):430-439.
doi pubmed - Bokenkamp A, Ariceta G, Bockenhauer D, Devuyst O, Emma F, van Bennekom D, Levtchenko E, et al. Dent disease: clinical practice recommendations. Nephrol Dial Transplant. 2025;40(5):852-864.
doi pubmed - Tosetto E, Ghiggeri GM, Emma F, Barbano G, Carrea A, Vezzoli G, Torregrossa R, et al. Phenotypic and genetic heterogeneity in Dent's disease—the results of an Italian collaborative study. Nephrol Dial Transplant. 2006;21(9):2452-2463.
doi pubmed - Ye Q, Shen Q, Rao J, Zhang A, Zheng B, Liu X, Shen Y, et al. Multicenter study of the clinical features and mutation gene spectrum of Chinese children with Dent disease. Clin Genet. 2020;97(3):407-417.
doi pubmed - van Berkel Y, Ludwig M, van Wijk JAE, Bokenkamp A. Proteinuria in Dent disease: a review of the literature. Pediatr Nephrol. 2017;32(10):1851-1859.
doi pubmed - Sakakibara N, Nagano C, Ishiko S, Horinouchi T, Yamamura T, Minamikawa S, Shima Y, et al. Comparison of clinical and genetic characteristics between Dent disease 1 and Dent disease 2. Pediatr Nephrol. 2020;35(12):2319-2326.
doi pubmed - Burballa C, Cantero-Recasens G, Prikhodina L, Lugani F, Schlingmann K, Ananin PV, Besouw M, et al. Clinical and genetic characteristics of Dent's disease type 1 in Europe. Nephrol Dial Transplant. 2023;38(6):1497-1507.
doi pubmed - Sekine T, Komoda F, Miura K, Takita J, Shimadzu M, Matsuyama T, Ashida A, et al. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant. 2014;29(2):376-384.
doi pubmed - Seye AI, Bauland C, Giraud H, Mechin V, Reymond M, Charcosset A, Moreau L. Quantitative trait loci mapping in hybrids between Dent and Flint maize multiparental populations reveals group-specific QTL for silage quality traits with variable pleiotropic effects on yield. Theor Appl Genet. 2019;132(5):1523-1542.
doi pubmed - Raina R, Chakraborty R, Sethi SK. Approach to children with isolated proteinuria. Clin Pediatr (Phila). 2021;60(9-10):407-415.
- Wu F, Cang Y, Zhou Y, et al. Genotype–phenotype analysis of CLCN5 variants in Dent disease. Pediatr Nephrol.2022;37(12):2929-2938.
- Gianesello L, Del Prete D, Anglani F, Calo LA. Genetics and phenotypic heterogeneity of Dent disease: the dark side of the moon. Hum Genet. 2021;140(3):401-421.
doi pubmed - Ishida Y, Nakano Y, Sakamoto T, Inoue Y, Tanaka T. 4-Phenylbutyrate improves low-molecular-weight proteinuria in a CLCN5 knock-in model. Int J Mol Sci. 2024;25(3):1264.
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, including commercial use, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.