| International Journal of Clinical Pediatrics, ISSN 1927-1255 print, 1927-1263 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Int J Clin Pediatr and Elmer Press Inc |
| Journal website https://ijcp.elmerpub.com |
Case Report
Volume 000, Number 000, April 2025, pages 000-000

Successful Treatment of COVID-19 and Multisystem Inflammatory Syndrome in Children Associated Severe Aplastic Anemia With Unrelated Donor Stem Cell Transplantation
Christopher J. Galleya, Shalini Sathib, Amy W. Davisc, Tara Sutherlanda, Orly R. Kleind, Bindu K. Sathie, f ![]()
aDepartment of Pediatrics, Valley Children’s Healthcare, Madera, CA, USA
bDepartment of Integrative Biology, University of California Berkeley, Berkeley, CA, USA
cDepartment of Pathology, Valley Children’s Healthcare, Madera, CA, USA
dDivision of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Palo Alto, CA, USA
eDepartment of Pediatric Hematology/Oncology, Valley Children’s Healthcare, Madera, CA, USA
fCorresponding Author: Bindu K. Sathi, Department of Pediatric Hematology/Oncology, Valley Children’s Healthcare, Madera, CA 93636, USA
Manuscript submitted September 19, 2024, accepted February 21, 2025, published online April 22, 2025
Short title: Treatment of MIS-C-Associated SAA With MUD HSCT
doi: https://doi.org/10.14740/ijcp550
| Abstract | ▴Top |
Multisystem inflammatory syndrome in children (MIS-C) due to COVID-19 has many known complications affecting different organ systems, including the hemopoietic system. Previous reports showed that children with severe aplastic anemia (SAA) secondary to MIS-C had different outcomes, some with spontaneous recovery of blood counts and others with refractory SAA. Another case described a young adult with paroxysmal nocturnal hemoglobinuria (PNH) who had refractory aplastic anemia from COVID-19. We describe an unusual case of SAA in a teenager with MIS-C who did not have spontaneous recovery of his blood cell counts, and he was ultimately treated with matched unrelated donor stem cell transplant. Next generation sequencing of bone marrow revealed a variant of unknown significance in the phosphatidylinositol glycan class A (PIG-A) gene. While further studies are needed, we hypothesize that this patient had subclinical PNH that was exacerbated by the acute COVID-19 infection and MIS-C. Stem cell transplantation can lead to successful outcome in this scenario.
Keywords: COVID-19; Multisystem inflammatory syndrome in children; Severe aplastic anemia; Transplantation
| Introduction | ▴Top |
COVID-19 complicated by multisystem inflammatory syndrome in children (MIS-C) is known to be associated with multisystem organ involvement along with hematological complications. This syndrome has similarities to Kawasaki disease, hemophagocytic lymphohistiocytosis, and toxic shock syndrome, with notable hematological findings of thrombocytopenia, lymphopenia with neutrophilic leukocytosis, and coagulopathy. Thrombocytopenia is the most common hematological symptom of MIS-C, but it is usually transient and resolves with supportive care.
Previous literature has shown pancytopenia as a known complication of MIS-C [1]; however, there are only two case reports of severe pancytopenia in an infant [2] and a child [3]. The former case showed transient pancytopenia that resolved after treatment of MIS-C with steroids while the latter case had persistent aplastic anemia that was refractory to antiviral therapy and immune modulators. Otieno et al reported on a 19-year-old female with undiagnosed paroxysmal nocturnal hemoglobinuria (PNH) who developed hemolysis and severe pancytopenia during an acute COVID-19 infection [4]. She had slight improvement of symptoms with eculizumab but had persistent pancytopenia and finally underwent bone marrow transplant evaluation.
Herein, we describe a teenager that had acute COVID-19 infection with MIS-C who developed severe aplastic anemia (SAA) and underwent successful treatment with matched unrelated donor (MUD) hematopoietic stem cell transplantation (HSCT) in June of 2022.
| Case Report | ▴Top |
Investigations
A 15-year-old male with a past medical history of asthma presented in December 2021 with 4 days of fever (102 - 103 °F), cough, sore throat, tremors, and fatigue. He had a seizure-like episode with whole body shaking, eye-rolling, and decreased arousal prior to arrival at the emergency room. A computed tomography (CT) scan did not reveal any intracranial pathology. At the time of admission to the hospital, he had elevated C-reactive protein (CRP), sedimentation rate, ferritin, fibrinogen, and D-dimers. There was a history of known sick contacts with COVID-19 at home. Physical exam revealed petechiae on the neck and pallor.
Diagnosis
Patient tested positive for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by nucleic acid amplification test (NAAT)/polymerase chain reaction (PCR) from the nasopharyngeal swab. Initial labs were significant for absolute neutrophil count (ANC) of 0.884 × 103/µL, platelets of 12 × 103/µL, and absolute reticulocyte count of 0.016 × 106/µL. ANC decreased further to 0.452 on subsequent labs. Inflammatory markers were elevated, including CRP (0.6 mg/dL), D-dimer (1.94 µg/mL), fibrinogen (569 mg/dL), and ferritin (519.6 ng/mL). Lactate dehydrogenase was normal (190 U/L). Peripheral smear showed macrocytic anemia with leukopenia and marked thrombocytopenia. B troponins and echocardiogram were normal. Echocardiogram revealed good ventricular function with normal strain indices and normal coronary arteries. There was no evidence of pericardial effusion. Due to elevation of inflammatory markers, seizures, and pancytopenia, a diagnosis of MIS-C was made. Bone marrow aspirate and biopsy was done due to persistence of pancytopenia. The biopsy revealed hypocellular bone marrow with 10% cellularity without dysplastic features in the bone marrow precursors (Figs. 1 and 2). Flow cytometry showed normal T- and B-cell populations without evidence of increased blasts. Chromosome analysis by fluorescent in situ hybridization showed a normal 46,XY karyotype with no translocations. Next generation sequencing revealed phosphatidylinositol glycan class A (PIG-A) c.524T>C, p.leu175Pro (11p15.5), a variant of unknown significance (VUS) with 5.3% variant frequency. There was no evidence of Philadelphia (Ph)-like or promyelocytic leukemia-retinoic acid receptor α (PML-RARα) translocation (Table 1).
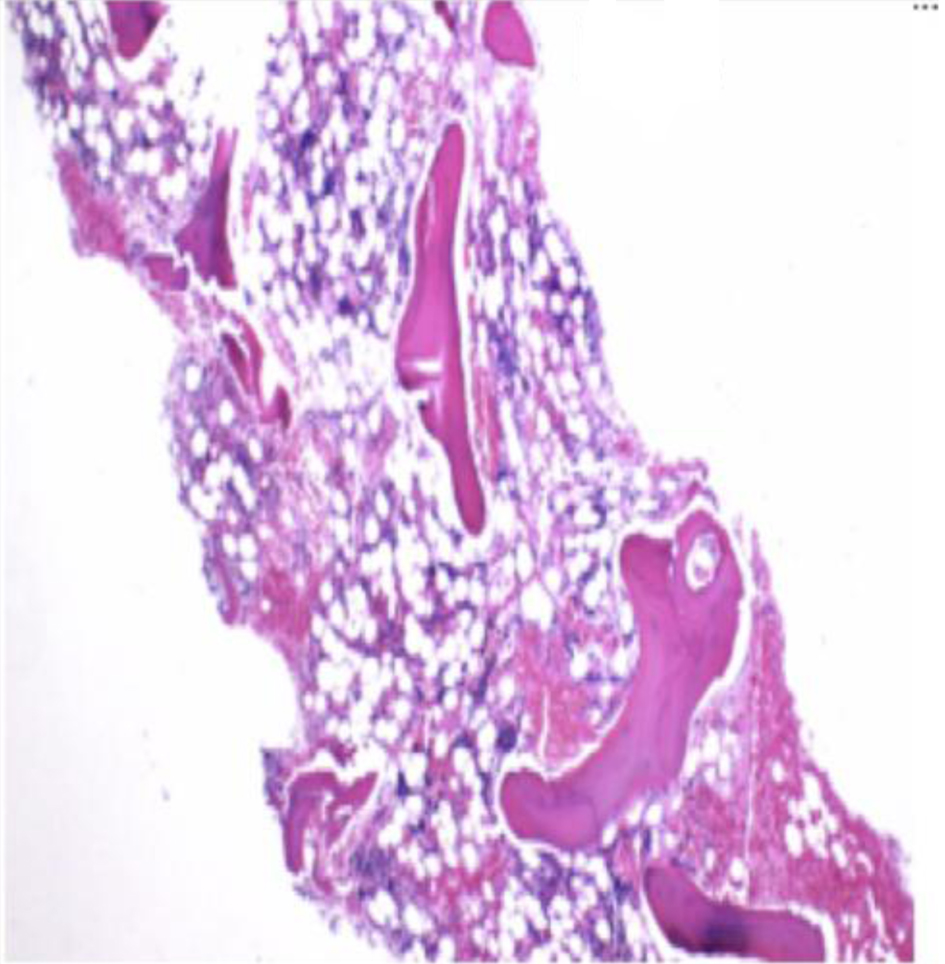 Click for large image | Figure 1. Bone marrow core biopsy at × 10. The core biopsy shows hypocellular marrow. Cellularity is approximately 10%. |
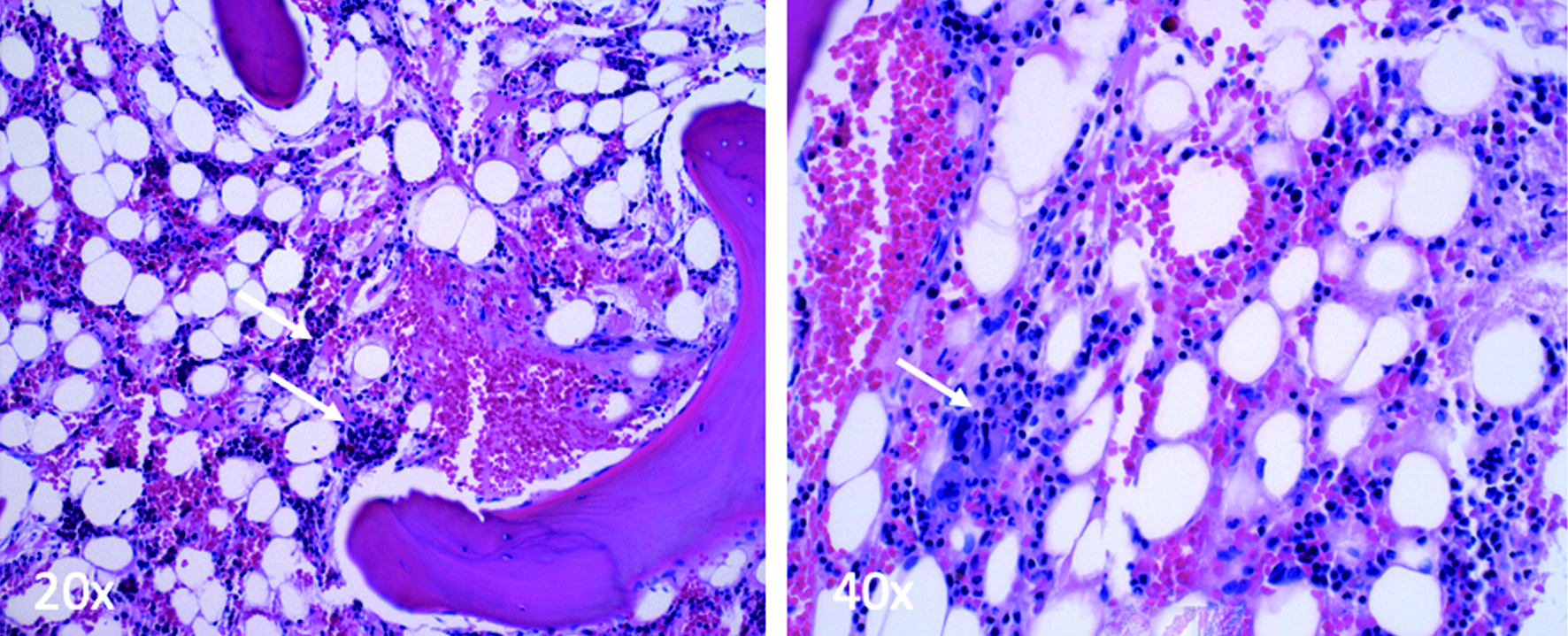 Click for large image | Figure 2. Small clusters of bone marrow precursors with trilineage hematopoiesis (white arrows) (× 20, × 40). No dysplastic elements or blast population are present. |
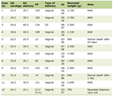 Click to view | Table 1. Workup for Aplastic Anemia |
Treatment
The patient was treated with intravenous immunoglobulin (IVIG) 115 g over 10 h and methylprednisolone 2 mg/kg/day twice daily due to clinical symptoms of MIS-C. Aminocaproic acid and oxymetazoline, in addition to platelet and packed red blood cell (pRBC) transfusion, were given due to persistent epistaxis and anemia. He was also started on a thrombopoietin receptor agonist, eltrombopag. However, significant improvement in blood count did not occur, and the decision was made to refer him for stem cell transplantation after discussion with his mother. Counts prior to transplantation were as follows: ANC of 0.419 × 103/µL, platelet count of 15 × 103/µL, and absolute reticulocyte count of 0.040 × 106/µL.
The patient required weekly pRBC and platelet transfusions as an outpatient over the next 4 months for symptomatic anemia and thrombocytopenia, respectively. He was also found to be vitamin D and zinc deficient (Table 1) and was supplemented. Prophylactic voriconazole and Bactrim were started due to persistent neutropenia. Both immunosuppressive therapy (IST) and stem cell transplantation options were discussed with family. IST consisted of rabbit anti-thymocyte globulin (r-ATG) and cyclosporine-A (Cs-A). The mother opted for stem cell transplantation as definitive therapy. As there was no human leukocyte antigen (HLA) matched sibling, unrelated donor search was initiated. He underwent HLA 10/10 MUD transplantation (research protocol BMT143, 146, 179, 328, 351 - Stanford University). The conditioning regimen consisted of r-ATG, fludarabine, and cyclophosphamide.
Post-transplantation course was complicated by grade 1 graft versus host disease (GVHD) of the skin. He was treated with prophylactic methotrexate, in addition to tacrolimus and steroids. Reactivation of latent cytomegalovirus infection occurred and was treated with valganciclovir.
Follow-up and outcome
Currently, he has completed 2 years post-transplantation and is transfusion-independent and fully immune-reconstituted.
| Discussion | ▴Top |
COVID-19 and MIS-C associated pancytopenia is generally transient, often resolving with supportive care. The subject was defined as having MIS-C due to involvement of the brain (seizures) and hematopoietic system. This case is unique with occurrence of pancytopenia refractory to steroids, IVIG, and eltrombopag. The subject required weekly platelet transfusions and was deemed a candidate for either HSCT or immunosuppressive therapy due to persistence of pancytopenia. We speculate that the trigger for the aplastic anemia and lack of recovery of counts could be in part due to MIS-C with co-existent PNH mutation, as a low-grade PNH clone was detected. The role of PNH mutation in aplastic anemia with COVID-19 is unknown but has been found in the context of aplastic anemia [1, 4].
Literature review was performed using PubMed and Google Scholar. Search terms included “(COVID-19 OR mis-c) AND aplastic anemia” and “COVID-19 AND mis-c AND aplastic.” Search results were selected to case reports with patients who developed pancytopenia or aplastic anemia following a COVID-19 infection, patients under 21 years of age, and in the English language. Patients with known aplastic anemia or other hematological conditions prior to acute COVID-19 infection were excluded. Seven articles met the above criteria, with results summarized in Table 2 [1-7].
Click to view | Table 2. Literature Review |
Literature review revealed two other published case reports of patients with subclinical PNH and resultant pancytopenia or aplastic anemia secondary to COVID-19 [1, 4]. One of the case reports showed a young adult female diagnosed with PNH during an acute COVID-19 infection; she also had persistent pancytopenia [4]. However, her laboratory workup showed some differences, such as erythroid predominant trilineage hematopoiesis on biopsy in the setting of active hemolysis. In contrast our subject did not have evidence of active hemolysis. A second patient [1] was also discovered to have PNH which was adequately treated with immunosuppressive therapy and biologics.
A close association between PNH and aplastic anemia has been known for nearly 50 years [8]; however, the full extent of this relationship is not fully understood. The most intriguing theory is that PNH clones become evident and expand after stress to normal cells. PNH clonal expansion is found in almost 50% of patients with immune-mediated acquired aplastic anemia [9]. It has even been reported after chloramphenicol use which had previously only been known for its association with aplastic anemia through both idiosyncratic and predictable bone marrow suppression [10]. It is unknown at this time if COVID-19 induces a similar form of stress to the stem cell and leads to expansion of PNH clone. PNH is an acquired somatic mutation in the PIG-A gene that results in failure to express the phosphatidylinositol-glycan anchor. This mutation leads to deficient expression of membrane proteins that ameliorate complement-mediated destruction of erythrocytes [11]. Interestingly, a correlation between SARS-CoV-2 infection and PNH appears to be emerging. In fact, from the beginning of the pandemic, many cases of clinical deterioration and increased hemolysis have occurred in patients with a known diagnosis of PNH following infection with COVID-19 [4, 12-18]. Moreover, if SARS-CoV-2 catalyzes an immunological attack against bone marrow progenitors, the virus appears to have the ability to cause PNH clonal expansion [9]. The proteins which fail to become anchored are therefore non-functional, including decay-accelerating factor (DAF, CD55), an inhibitor of alternative pathway C3 convertase, and protectin (CD59), an inhibitor of membrane attack complex (MAC) formation. It has been hypothesized that SARS-CoV-2 may be involved in the direct transcription of pro-inflammatory mediators in hematopoietic stem cells (HSCs) by a spike protein interaction with the angiotensin-converting enzyme 2 (ACE2) receptor. It may also induce uncontrolled NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome expression, leading to HSC death via pyroptosis [19]. Alternatively, the pro-inflammatory microenvironment generated by COVID-19 may also play a mechanistic role in the immune-mediated acquired aplastic anemia. The development of abnormal autoimmune responses, including cytotoxic T cells that activate, expand, and circulate as oligoclones, causes the release of myelosuppressive cytokines and induces the cell death of HSCs and progenitor cells [19]. Whether the SARS-CoV-2 virus can act as an inciting antigen remains to be clarified, but what is already known is that the presence of PNH clones irrespective of size is a good predictor of response to immunosuppressive therapy and stem cell transplant [9].
Aside from comorbid hematopoietic disorders such as PNH, several viral infections are known to cause SAA in both children and adults [5-7, 20, 21]. COVID-19 associated aplastic anemia in children was treated by immunosuppressive therapy or observation with supportive care [11-13]. Acute COVID-19 infection itself and its effect on HSCs may also have contributed to this patient’s anemia. Further studies are needed to elucidate the full molecular and immunological mechanism underlying this presentation.
Learning points
This case describes SAA as a novel complication of COVID-19 associated MIS-C. The pathogenesis of this presentation is not yet fully elucidated, but underlying PNH clone could be contributing to the severity of symptoms. Further research is needed to explore the role of COVID-19 MIS-C on exacerbation of co-morbid hematopoietic conditions.
Acknowledgments
We acknowledge Brian Baker for assistance with literature review.
Financial Disclosure
Bindu K. Sathi is a consultant with Vertex Pharmaceuticals. No other financial disclosures to disclose.
Conflict of Interest
None to declare.
Informed Consent
Informed consent was obtained from the mother of the subject.
Author Contributions
Christopher Galley wrote the initial draft. Shalini Sathi wrote and edited the draft. Tara Sutherland and Orly Klien treated the patient and edited the draft. Amy Davis evaluated the marrow aspirates and biopsies and edited the draft. Bindu K. Sathi treated the patient, conceptualized, wrote, and finalized the draft.
Data Availability
The data supporting the findings of the study are available from corresponding author upon reasonable request.
Abbreviations
ANC: absolute neutrophil count; Cs-A: cyclosporine-A; GVHD: graft versus host disease; HLA: human leukocyte antigen; HSCT: hematopoietic stem cell transplantation; IST: immunosuppressive therapy; IVIG: intravenous immunoglobin; MIS-C: multisystem inflammatory syndrome in children; MUD: matched unrelated donor; NAAT: nucleic acid amplification test; NLRP3: NOD-, LRR-, and pyrin domain-containing protein 3; PCR: polymerase chain reaction; PIG-A: phosphatidylinositol glycan class A; PML-RARα: promyelocytic leukemia-retinoic acid receptor α; PNH: paroxysmal nocturnal hemoglobinuria; pRBC: packed red blood cell; r-ATG: rabbit anti-thymocyte globulin; SAA: severe aplastic anemia; VUS: variant of unknown significance
| References | ▴Top |
- Lee NCJ, Patel B, Etra A, Bat T, Ibrahim IF, Vusirikala M, Chen M, et al. SARS-CoV-2 infection associated with aplastic anemia and pure red cell aplasia. Blood Adv. 2022;6(13):3840-3843.
doi pubmed - Mariani R, Liu H. Severe transient pancytopenia with dyserythropoiesis and dysmegakaryopoiesis in COVID-19-associated MIS-C. Blood. 2020;136(25):2964.
doi pubmed - Figlerowicz M, Mania A, Lubarski K, Lewandowska Z, Sluzewski W, Derwich K, Wachowiak J, et al. First case of convalescent plasma transfusion in a child with COVID-19-associated severe aplastic anemia. Transfus Apher Sci. 2020;59(5):102866.
doi pubmed - Otieno SB, Altahan A, Kaweeta F, Karri S, Alnoor F, Johnson R. Severe hemolysis in a COVID-19 patient with paroxysmal nocturnal hemoglobinuria. Case Rep Hematol. 2021;2021:6619177.
doi pubmed - Amanati A, Hedayati SB, Ziyaeyan M, Honar A, Dashtianeh R, Rabiei N, Saki N, et al. A fatal SARS-coronavirus-2 induced bone marrow aplasia complicated with invasive fungal infection and severe neutropenic enterocolitis. BMC Infect Dis. 2022;22(1):682.
doi pubmed - Chakravarthy R, Murphy ML, Ann Thompson M, McDaniel HL, Zarnegar-Lumley S, Borinstein SC. SARS-CoV-2 infection coincident with newly diagnosed severe aplastic anemia: A report of two cases. Pediatr Blood Cancer. 2022;69(4):e29433.
doi pubmed - Ranjima M. Severe aplastic anemia secondary to SARS CoV-2 infection- a case report. Journal of Pediatrics, Perinatology and Child Health Journal of Pediatrics, Perinatology and Child Health. 2021;5:230-237.
- Dacie JV, Lewis SM. Paroxysmal nocturnal haemoglobinuria: variation in clinical severity and association with bone-marrow hypoplasia. Br J Haematol. 1961;7:442-457.
doi pubmed - Fattizzo B, Ireland R, Dunlop A, Yallop D, Kassam S, Large J, Gandhi S, et al. Clinical and prognostic significance of small paroxysmal nocturnal hemoglobinuria clones in myelodysplastic syndrome and aplastic anemia. Leukemia. 2021;35(11):3223-3231.
doi pubmed - Diskin C. Paroxysmal nocturnal hemoglobinuria after chloramphenicol therapy. Mayo Clin Proc. 2005;80(10):1394.
doi pubmed - Takahashi M, Takeda J, Hirose S, Hyman R, Inoue N, Miyata T, Ueda E, et al. Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuria. J Exp Med. 1993;177(2):517-521.
doi pubmed - Kulasekararaj AG, Lazana I, Large J, Posadas K, Eagleton H, Lord Villajin J, Zuckerman M, et al. Terminal complement inhibition dampens the inflammation during COVID-19. Br J Haematol. 2020;190(3):e141-e143.
doi pubmed - Genthon A, Chiarabini T, Baylac P, Valin N, Urbina T, Pacanowski J, Mekinian A, et al. Severe COVID-19 infection in a patient with paroxysmal nocturnal hemoglobinuria on eculizumab therapy. Leuk Lymphoma. 2021;62(6):1502-1505.
doi pubmed - Fattizzo B, Pasquale R, Bellani V, Barcellini W, Kulasekararaj AG. Complement mediated hemolytic anemias in the COVID-19 era: case series and review of the literature. Front Immunol. 2021;12:791429.
doi pubmed - Pike A, Muus P, Munir T, Mitchell L, Arnold L, Riley K, Houghton N, et al. COVID-19 infection in patients on anti-complement therapy: The Leeds National Paroxysmal Nocturnal Haemoglobinuria service experience. Br J Haematol. 2020;191(1):e1-e4.
doi pubmed - Schuller H, Klein F, Lubbert M, Prager EP. Hemolytic crisis in a patient treated with eculizumab for paroxysmal nocturnal hemoglobinuria possibly triggered by SARS-CoV-2 (COVID-19): a case report. Ann Hematol. 2021;100(3):841-842.
doi pubmed - Sokol J, Nehaj F, Mokan M, Lisa L, Stasko J. COVID19 infection in a patient with paroxysmal nocturnal hemoglobinuria: A case report. Medicine (Baltimore). 2021;100(20):e25456.
doi pubmed - Aguilar JJ, Dhillon V, Balasubramanian S. Manifestation of pancytopenia associated with COVID-19 as Paroxysmal Nocturnal Hemoglobinuria (PNH) and Aplastic Anemia (AA). Hematol Rep. 2024;16(1):42-49.
doi pubmed - Zeng Y, Katsanis E. The complex pathophysiology of acquired aplastic anaemia. Clin Exp Immunol. 2015;180(3):361-370.
doi pubmed - Miano M, Dufour C. The diagnosis and treatment of aplastic anemia: a review. Int J Hematol. 2015;101(6):527-535.
doi pubmed - Kurre P, Johnson FL, Deeg HJ. Diagnosis and treatment of children with aplastic anemia. Pediatr Blood Cancer. 2005;45(6):770-780.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
International Journal of Clinical Pediatrics is published by Elmer Press Inc.